Avaliação de risco como ferramenta de garantia de qualidade de processo
Por Marco Casteldo

O Conceito de Risco na Produção Farmacêutica
No rascunho divulgado do Anexo 1 das BPF da UE, Revisão 12, destaca-se a importância do gerenciamento de riscos (Gerenciamento de Riscos) como uma ferramenta adequada para garantir a qualidade de um processo. O rascunho é explícito sobre a necessidade de gerenciamento de risco para fabricantes de medicamentos estéreis e também recomenda amplamente o gerenciamento de risco para outros tipos de produtos, especialmente quando o controle de contaminação microbiana, por partículas e pirogênio é necessário (por exemplo, certos líquidos, cremes, pomadas, e intermediários bacterianos baixos) [1].
A produção e uso de um medicamento (medicamento) e seus componentes envolvem necessariamente um certo grau de risco. É importante entender que a qualidade do produto deve ser mantida durante todo o ciclo de vida do produto. Isso garante que os atributos importantes para a qualidade do medicamento (Atributos de Qualidade Críticos) permaneçam consistentes ao longo das fases de desenvolvimento e produção do medicamento. De acordo com a ICH Q6A, a qualidade do medicamento refere-se à adequação de um medicamento ou medicamento para o uso pretendido [5]. Em geral, um procedimento de gerenciamento de risco concentra-se na análise de cada processo em um ciclo de vida do produto com a intenção de realizar uma avaliação, mitigação e revisão dos riscos associados ao longo do tempo. Conforme definido no ICH Q9,
Quando se fala em qualidade farmacêutica, o termo “processo” pode assumir diferentes significados. Pode referir-se a qualquer um dos estágios de desenvolvimento, produção, teste, inspeção, distribuição, até e incluindo a entrega de medicamentos. Além disso, pode incluir o projeto, qualificação e validação de equipamentos, instrumentos e instalações. Assim, um processo é qualquer atividade que possa afetar direta ou indiretamente a qualidade do produto final. É imediatamente aparente que o escopo da gestão de risco de qualidade farmacêutica é muito amplo.
Antes de aprofundar os procedimentos de gerenciamento de riscos, é bom refletir sobre algumas distinções importantes, começando com a diferença entre perigos e problemas. Os problemas estão relacionados à percepção ou implementação de um processo [4], enquanto um perigo é entendido como uma propriedade ou qualidade intrínseca que tem o potencial de causar danos ao processo e, portanto, ao cliente final.
Portanto, o risco é um conceito probabilístico; é a combinação da probabilidade de um determinado evento ocorrer e a capacidade desse evento de causar danos. Isso é ainda mais complicado pela incapacidade de detectar esse risco no momento da ocorrência. Do ponto de vista técnico, o risco é o produto da probabilidade e da gravidade [2], onde a detectabilidade - se introduzida - deve considerar o risco de falha do sistema de detecção. A noção de risco implica, assim, a existência de uma fonte de perigo e a possibilidade de se transformar em dano [3].
É a partir deste momento na análise da Gestão de Riscos que o risco é efetivamente atualizado e traduzido em algo científico e documentado. A partir deste ponto, os testes e avaliações da avaliação de risco devem ser transformados em algo visual e quantitativo para que tanto a capacidade de gerenciar o risco quanto a origem do risco possam ser compreendidas externamente. A ferramenta final de avaliação de risco é a identificação de perigos e a análise e avaliação científica dos riscos associados à exposição a eles. Isso inclui a gravidade dos danos ou danos à saúde, incluindo danos logísticos que podem resultar da perda de qualidade ou disponibilidade do produto.
É essencial ter um sistema de gestão de qualidade robusto e boas práticas de fabricação para reduzir os riscos à qualidade do produto, segurança do paciente e reputação da empresa a um nível aceitável.
Existem dois princípios básicos de gerenciamento de riscos de qualidade:
- As avaliações de risco devem ser baseadas no conhecimento científico e orientadas pela proteção do paciente.
- O nível de comprometimento, formalidade e documentação do processo de gerenciamento de riscos de qualidade deve ser proporcional ao nível de risco.
Avaliação de risco 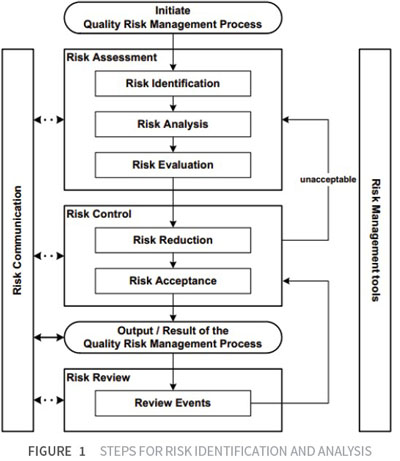
O processo de avaliação de riscos consiste em diferentes etapas que vão desde a identificação dos perigos até a análise e avaliação dos riscos associados à exposição a esses perigos. A Figura 1 mostra as etapas desenvolvidas durante a análise.
A etapa de identificação de riscos é composta pelo uso sistemático de informações para identificar potenciais fontes de danos (perigos) e possíveis consequências (impacto/efeito). A identificação desses danos e consequências é baseada em dados históricos, análises teóricas, opiniões informadas, preocupações das partes interessadas, sessões de brainstorming, etc. Essas informações são necessárias para aprofundar o conhecimento dos processos.
Essas três perguntas básicas costumam ser úteis como auxílio para definir claramente o risco:
- O que poderia ser diferente do esperado?
- Qual é a probabilidade (probabilidade) de ser diferente da expectativa?
- Quais são as consequências (gravidade)?
A análise de risco é a estimativa do risco associado aos perigos identificados. É um processo qualitativo ou quantitativo de vincular a probabilidade e a gravidade do dano (sendo a gravidade uma medida das possíveis consequências de um perigo) avaliando o projeto/medidas que têm controle sobre sua ocorrência e detecção [2]. Analisar o grau de risco leva à definição de ferramentas ou ações adequadas para sua gestão ao longo do tempo. Com algumas ferramentas de gerenciamento de risco, a capacidade de detectar danos (detectabilidade) também pode ser considerada um fator que influencia a estimativa geral de risco.
A avaliação de risco leva então à comparação do risco estimado com determinados critérios de risco. Isso é feito por meio de uma escala quantitativa ou qualitativa que determina sua significância e, posteriormente, define um limite de aceitabilidade. Quando o risco é expresso quantitativamente, uma probabilidade numérica é usada. Alternativamente, o risco pode ser expresso usando descritores qualitativos, como "alto", "médio" ou "baixo", que devem ser definidos com o máximo de detalhes possível. O objetivo do controle de risco é reduzir o risco a um nível aceitável e definido.
Portanto, a avaliação deve levar a uma aceitação do próprio risco (controle de risco), se o nível for aceitável, ou a uma redução do risco, se não for um nível aceitável.
O processo de controle de risco pode incluir ações para:
- diminuir a probabilidade de ocorrência dos perigos e riscos identificados
- diminuir a gravidade dos perigos e riscos identificados
- aumentar a detectabilidade dos perigos e riscos identificados
Deve-se sempre ter em mente que a implementação de medidas de redução de risco pode introduzir novos riscos no sistema (risco induzido) ou aumentar a significância de outros riscos já existentes (risco correlacionado). Portanto, pode ser apropriado revisar a avaliação após a implementação de um processo de redução de risco para identificar e avaliar possíveis mudanças. A frequência de qualquer revisão deve ser baseada no nível de risco. A revisão de risco pode incluir a reconsideração das decisões de aceitação de risco [2, 4].
A aceitação só é possível quando comprovado cientificamente que a qualidade final do processo não é impactada criticamente pelo risco identificado ou residual. Para alguns tipos de danos, mesmo as melhores práticas de gerenciamento de risco de qualidade podem não eliminar todo o risco. Nessas circunstâncias, a aplicação de uma estratégia de gerenciamento de risco apropriada reduz o risco à qualidade a um nível especificado (aceitável). Este nível aceitável dependerá de muitos parâmetros e deve ser decidido caso a caso. Vários processos, incluindo uma análise de custo-benefício, podem ser usados para entender o nível ótimo de controle de risco, sempre em conformidade com os requisitos regulatórios e normativos [2].
Gestão de Riscos e Possíveis Abordagens
Vários métodos científicos estão atualmente disponíveis para uso na análise de risco. Estes podem ser usados independentemente ou em conjunto uns com os outros para alcançar os resultados mais construtivos. Por exemplo, usar uma combinação do método HACCP com FMEA pode levar à produção de documentos mais abrangentes do que qualquer um por si só. Cobrimos três deles abaixo.
APPCC
A abordagem HACCP é uma ferramenta sistemática, proativa e preventiva para garantir a qualidade, confiabilidade e segurança do produto [7]. Com base no desenvolvimento de uma “Árvore de Decisão”, a abordagem HACCP facilita a identificação de áreas críticas e não críticas do processo em análise.
FMEA
A abordagem FMEA é uma análise sistemática de modos de falha potenciais com o objetivo de prevenir falhas. É um processo de ação preventiva implementado antes da introdução de novos produtos, modificações ou processos. Idealmente, as análises FMEA são realizadas nas fases de projeto ou desenvolvimento do processo/produto, no entanto, também podem ser muito úteis quando aplicadas a produtos e processos existentes. Esta abordagem é aplicável em uma variedade de áreas, incluindo a fabricação e montagem farmacêutica. Consiste em várias etapas que incluem a revisão do processo, identificação de modos de erro potenciais, listagem dos efeitos potenciais de cada modo de erro, atribuição de gravidade/ocorrência e detecção para cada efeito e cálculo do Número de Prioridade de Risco (RPN) para priorizar ações de mitigação que eliminem ou reduzir o risco.
Número de prioridade de risco (RPN)
O cálculo do Número de Prioridade do Risco representa o resultado obtido a partir da avaliação de três parâmetros: "Gravidade" (a gravidade do risco), "Detecção" (a capacidade de detectar o risco) e "Ocorrência" (a probabilidade de que o risco possa ocorrer e se repetir dentro do processo em análise). Para cada um desses parâmetros, as variáveis são avaliadas e depois multiplicadas para obter a avaliação final. (RPN = Gravidade x Detecção x Ocorrência) [6]. Uma vez estabelecidas as variáveis para cada parâmetro individual, as pontuações máxima e mínima podem ser calculadas para definir os intervalos. Um valor de risco de "baixo" seria atribuído se o valor da pontuação estiver dentro do intervalo mais baixo, "médio" se o valor da pontuação estiver dentro do intervalo intermediário e "alto" se o valor RPN estiver dentro do intervalo mais alto. A divisão em diferentes faixas de risco é necessária para a conclusão da próxima etapa de controle de risco. Atribuir um valor de risco a uma faixa em vez de outra contribui para o processo de tomada de decisão de redução ou aceitação de risco. Se o risco total identificado for médio ou alto, então ações corretivas ou preventivas de controle de risco devem ser aplicadas ao processo para diminuir seu valor (ou seja, risco de impacto na qualidade).
Além disso, sempre que possível, o valor do risco deve ser reduzido a um valor limite aceitável. Estar na faixa "baixa" determina a aceitação do risco como estando abaixo de um limite aceitável estabelecido e não exigindo ação corretiva ou preventiva porque já está sob controle. Se tomarmos como exemplo o envase e o acabamento de um produto, uma boa estratégia de prevenção da contaminação ambiental do processo de produção estéril torna-se essencial para garantir a qualidade do produto acabado.
A avaliação de risco é, portanto, uma ferramenta necessária e fundamental para as empresas farmacêuticas utilizarem para fortalecer o processo de garantia de qualidade, aliada à instrumentação utilizada para realizar praticamente o que é avaliado teoricamente. A boa historicização dos dados e a representação adequada dos dados podem ser parâmetros a serem considerados para facilitar o controle de risco e o processo de revisão. O uso apropriado da gestão de riscos de qualidade pode facilitar (mas não elimina) a obrigação da indústria de cumprir os requisitos regulatórios [2].
Conclusões
Uma abordagem eficaz ao gerenciamento de risco pode garantir ainda mais a entrega de um medicamento ou medicamento de alta qualidade ao paciente, fornecendo um meio proativo para identificar e controlar possíveis problemas de qualidade durante o desenvolvimento e a fabricação de medicamentos. O principal objetivo de qualquer gestão de risco deve ser a proteção do usuário final do produto, e a qualidade do produto é a medida definitiva do sucesso de uma estratégia de risco de qualidade que identifica e mantém a segurança do cliente final. Uma avaliação de risco totalmente documentada permite que as empresas farmacêuticas obtenham produtos de alta qualidade e cumpram as diretrizes e requisitos regulatórios.
A gestão contínua da qualidade do produto é assegurada através do controlo microbiológico e de partículas do ar e das superfícies de acordo com um plano de monitorização definido. Também é importante ter em mente a interdependência das abordagens de gestão de risco e avaliação de risco. Pode ser útil utilizar organizações externas para orientação. Esses especialistas podem ajudar a empresa a definir e gerenciar seus riscos compartilhando informações coletadas de forma clara e sistemática. A comunicação pode ocorrer em qualquer ponto do processo e as informações compartilhadas podem dizer respeito à existência, natureza, forma, probabilidade, gravidade, aceitabilidade, controle, tratamento, detectabilidade ou outros aspectos dos riscos de qualidade, conforme necessário.
Referências
[1] EudraLex - Volume 4 - Diretrizes de Boas Práticas de Fabricação (BPF) – Anexo I
[2] ICH Q9 Gestão do risco de qualidade
[3] art. 2, letra s, D. Lgs. 81/08
[4] Guia ISO/IEC 51:1999 - Aspectos de Segurança - Diretriz para sua inclusão em padrões
[5] ICH Q6A
[6] Noções básicas de FMEA, Robin McDermott, Raymond J. Mikulak, Michael R. Beauregard 1996, ISBN 0527763209.
[7] Série de Relatórios Técnicos da OMS Nº 908, 2003 Anexo 7
Sobre o autor
Marco Castaldo, chefe da equipe de consultoria, Particle Measuring Systems 
Marco tem uma formação diversificada na área farmacêutica, incluindo experiência nas áreas de Garantia de Esterilidade, Garantia de Qualidade e Conformidade. Adquiriu ampla experiência em grandes empresas multinacionais antes de se tornar Consultor e Gerente de Projetos. Marco auxilia empresas farmacêuticas em projetos que incluem: validação de novas tecnologias, redução de contaminação microbiológica e auditorias internas para comportamentos assépticos. Como chefe da equipe consultiva do sistema de medição de partículas, ele está focado em apoiar empresas farmacêuticas em todo o mundo para melhorar sua abordagem e estratégia de garantia de esterilidade.
Nenhum comentário:
Postar um comentário