NMR e espectrometria de massa em desenvolvimento farmacêutico
Por Joshua Hicks, Ph.D., Cientista Sênior, Espectroscopia Orgânica, Catalent

A espectroscopia de ressonância magnética nuclear (NMR) e a espectrometria de massa (MS) são técnicas poderosas para a caracterização e quantificação de compostos. Suas aplicações incluem a capacidade de confirmar a estrutura dos compostos, determinar a pureza e identificar (e possivelmente quantificar) impurezas relacionadas ao processo, degradantes ou outras incógnitas observadas durante a triagem por cromatografia líquida. Juntas, as análises de NMR e MS podem identificar definitivamente os padrões de referência, ingredientes farmacêuticos ativos (APIs) ou a composição final do medicamento. Essas técnicas podem ser especialmente úteis para identificar constituintes desconhecidos em uma amostra e / ou confirmar a identidade de materiais onde não há padrões de referência disponíveis. Neste artigo, a capacidade de NMR e MS para caracterizar a composição do produto é demonstrada usando exemplos detalhados.
NMR PARA CARACTERIZAÇÃO DE UM PADRÃO DE REFERÊNCIA
Os padrões de referência são amplamente usados para estabelecer identidade, qualidade, potência e pureza de substâncias para uso farmacêutico. Eles são usados nas várias fases do desenvolvimento de medicamentos. Para atender aos padrões das agências reguladoras, os padrões de referência devem ser bem caracterizados para uso na confirmação da identidade das substâncias durante os testes analíticos.
Dependendo da fonte, os padrões de referência podem ser considerados materiais de referência primários ou materiais de referência secundários.
- Os materiais de referência primários (PRM) são compostos totalmente caracterizados de alta pureza usados para ensaios, pureza ou testes de identificação. Eles são tipicamente obtidos de farmacopeias como a Farmacopeia dos Estados Unidos (USP) e a Farmacopeia Europeia (EP). Esses padrões são avaliados com base em um órgão de normalização nacional ou internacional. Quando um PRM não estiver disponível na USP ou EP, o patrocinador deve estabelecer o PRM por meio da caracterização apropriada dos materiais disponíveis.
- Os materiais de referência secundários (ou padrões de trabalho) também são de alto nível, mas foram qualificados em relação a um material de referência primário. Esses materiais são obtidos de outras fontes, incluindo fabricante contratado, fornecedor de produtos químicos ou sintetizados internamente.
Recentemente, tem havido muito mais escrutínio quando se trata de padrões de referência obtidos de "outras fontes". Não se pode mais confiar, por exemplo, na espectroscopia de infravermelho com transformada de Fourier (FTIR) ou apenas em Karl Fischer; São necessárias técnicas de caracterização mais rigorosas que sejam mais definitivas e possam identificar purezas com muito mais certeza para demonstrar a adequação do padrão de referência para o propósito pretendido.
É importante ter em mente os seguintes trechos da Food and Drug Administration (FDA) e EP:
“Os padrões de referência que não são obtidos de fontes oficiais ... devem ser completamente caracterizados para garantir sua identidade, força, qualidade, pureza e potência.”
Orientação da FDA "Validação de Métodos e Procedimentos Analíticos"
“Padrão primário Uma substância ou preparação a ser estabelecida por uma variedade de técnicas analíticas escolhidas para demonstrar sua adequação para uso.”
Padrões de Referência da Farmacopeia Europeia
NMR PARA CARACTERIZAÇÃO DE UMA ESTRUTURA QUÍMICA
Usando NMR, a caracterização do padrão de referência é realizada em diferentes níveis:
- Impressão digital do perfil espectral de NMR para comparação com o material de referência.
- Atribuição de deslocamento químico de átomos dentro de espectros de NMR para caracterização / confirmação estrutural.
- Atribuições de deslocamento químico e atribuições de estrutura secundária para pequenas proteínas e peptídeos.
- Cálculos de estrutura completa da estrutura terciária para pequenas proteínas e peptídeos.
O poder de usar NMR e padrões de referência para demonstrar a mesma configuração, conformação e identidade de substâncias é tão simples quanto comparar os espectros de NMR, seja uma molécula pequena ou macromolécula. A caracterização por NMR é abrangente e definitiva porque todas as informações estão presentes. Embora o espectro gerado possa ser muito complexo, os resultados são altamente reproduzíveis.
Identificando uma proteína
A Figura 1 mostra um exemplo de um espectro de caracterização de NMR utilizando espectroscopia de correlação total de coerência quântica única heteronuclear (HSQC-TOCSY) para identificar resíduos de aminoácidos individuais em um peptídeo. A identidade de aminoácidos individuais é determinada pela correlação entre as atribuições de deslocamento químico do carbono e do próton de cada resíduo de aminoácido. No eixo x estão os deslocamentos químicos para prótons e no eixo y estão os deslocamentos químicos para carbonos. Como exemplo, a linha roxa inferior representa um dos deslocamentos químicos do carbono (eixo y) de um resíduo de prolina, e cada cruzamento do contorno do eixo x (também conhecido como pico cruzado) representa prótons específicos na prolina. Um padrão específico é gerado por cada aminoácido encontrado em qualquer proteína ou peptídeo.
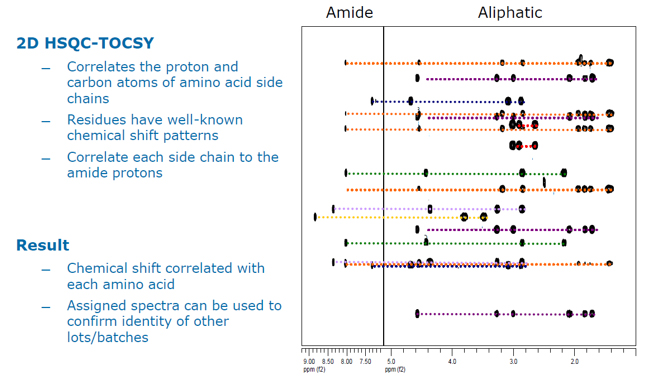
As mudanças químicas também podem ser usadas para confirmar estruturas secundárias, terciárias e quaternárias de peptídeos, pois as interações que criam uma conformação 3D específica de uma proteína levam a mudanças químicas no espectro de NMR exclusivo dessa proteína quando ela está em uma determinada conformação. Assim, se os espectros de uma amostra são comparados com os espectros de outra amostra ou um padrão de referência, a similaridade dos perfis espectrais é usada para confirmar que o mesmo material está presente em ambas as amostras.
Identificando um excipiente
O NMR também pode ser aplicado para a confirmação da identidade de excipientes usados em formulações farmacêuticas. Por exemplo, os materiais comumente testados incluem poloxâmeros, usados em muitas indústrias, incluindo produtos farmacêuticos e cosméticos. Um poloxâmero é um copolímero tribloco feito de um componente hidrofílico (A) e um componente hidrofóbico (B) em um arranjo ABA (Figura 2). As propriedades e o tipo de um poloxamer são determinados pelo tamanho das unidades A e B na cadeia do polímero. Os diferentes tipos incluem poloxâmero 124, 188, 237, 338 e 407. Os poloxâmeros são usados, por exemplo, para ligar um medicamento ou estabilizar cores de maquiagem ou bases.
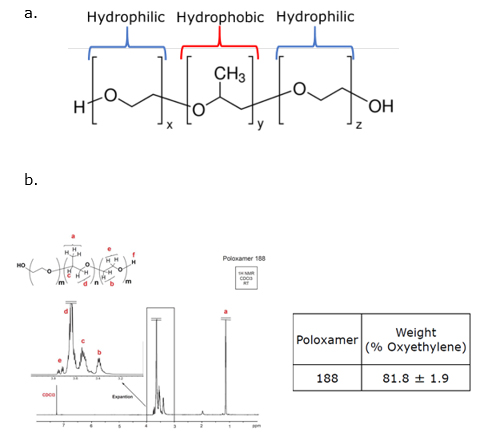
Aproveitando o NMR e sua reprodutibilidade, as monografias gerais do EP e da USP fazem referência a alterações químicas específicas para poloxâmeros. A Figura 2 mostra que os prótons individuais são identificados (por exemplo, B vai para B, A vai para A) no espectro obtido durante a análise de NMR. Dependendo do poloxamer, a área sob a região de deslocamento químico mudará e está diretamente correlacionada ao número de prótons encontrados na região integral.
Ao completar uma caracterização completa de um poloxâmero, uma única medição espectral de NMR pode identificar e quantificar o poloxâmero dentro de uma amostra. A identidade do poloxâmero é revelada pela porcentagem em peso de oxietileno, que é calculada por uma fórmula fornecida pela USP (EP se refere a isso como uma proporção, mas é a mesma fórmula). A quantidade do poloxâmero pode ser identificada pela intensidade dos picos dos espectros. Neste caso, o peso percentual de oxietileno é 81,8%, o que confirma a identidade como poloxâmero 188. NMR tem a capacidade de identificar (% oxietileno) e quantificar (intensidade de pico) em uma amostra de poloxâmero com uma medição espectral.
Identificando uma API genérica
Outra área que aproveita a caracterização estrutural de NMR é o maior rigor exercido pelo FDA para a demonstração de equivalência terapêutica em genéricos. Os fabricantes de genéricos precisam demonstrar que seu API, seja um peptídeo, proteína ou mesmo ácido nucleico, tem o mesmo perfil de segurança e propriedades de efeito clínico que o medicamento listado de referência. NMR é uma técnica definitiva para este teste por causa de sua linearidade inerente e reprodutibilidade aplicada a uma comparação direta entre perfis espectrais.
NMR PARA DETERMINAR PUREZA ABSOLUTA
O papel mais comum para NMR em estudos de quantificação é determinar a pureza absoluta. Cada vez que um espectro para um determinado composto é coletado, o mesmo deslocamento químico é relatado e a área sob o pico se correlaciona diretamente com a quantidade de composto na amostra. O espectro da amostra pode ser comparado ao de um padrão interno para determinar a pureza da amostra.
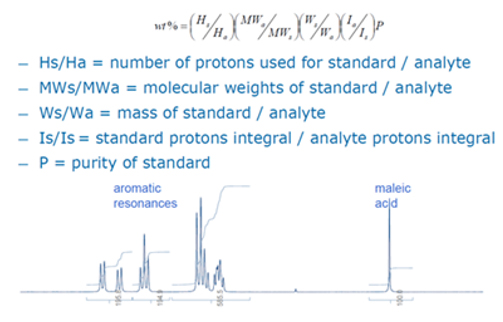
A Figura 3 mostra a aplicação de quantificação de um composto (isto é, um peptídeo) por uma comparação da área do pico com um padrão de referência interno de uma pureza conhecida. O padrão de referência, neste caso o ácido maleico, é um padrão certificado pelo NIST altamente caracterizado com pureza de 99%. A pureza do peptídeo é calculada relacionando diretamente a área da curva do ácido maleico com a área da curva do peptídeo usando a equação mostrada na Figura 3. Esses ensaios são realizados com frequência, uma vez que o método foi verificado para sua aplicação pretendida .
ESPECTROMETRIA DE MASSA IDENTIFICA UMA API
A espectrometria de massa é uma ferramenta muito sensível e apenas uma pequena amostra é necessária. A espectrometria de massa é usada para caracterização de padrão de referência por massa nominal ou precisa e para conduzir análises de fragmentação, como procurar modificações em diferentes fragmentos de compostos. Geralmente, é usado para identificar compostos com base em métodos verificados ou validados. Assim como o NMR, a espectrometria de massa pode ser usada para monitorar a estabilidade, quantificar os componentes principais e secundários e monitorar e quantificar os degradantes.
Para demonstrar a utilidade do MS na identificação de um composto, os cientistas da Catalent conduziram uma investigação cega da composição do comprimido. Vários sites Catalent receberam um comprimido de composição desconhecida e a tarefa de identificar e quantificar o API dentro do comprimido.
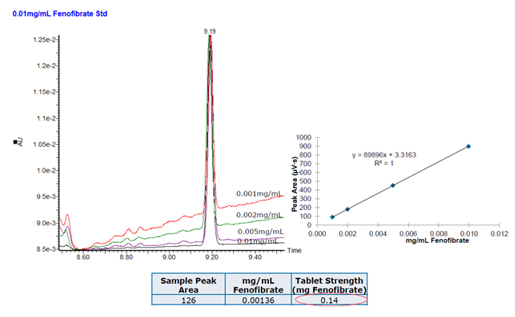
Na Figura 4, o painel esquerdo (retângulo azul) mostra um pico no traço UV. Os resultados do cromatograma de íons extraídos (XIC) e do cromatograma de íons total (TIC) foram usados para determinar a massa precisa. Com a massa precisa, foi possível restringir possíveis fórmulas empíricas. Uma avaliação de dados adicional revelou vários candidatos à fórmula; apenas alguns eram plausíveis. Seguindo essa suposição, os compostos de referência candidatos foram garantidos e testados, procurando uma correspondência.
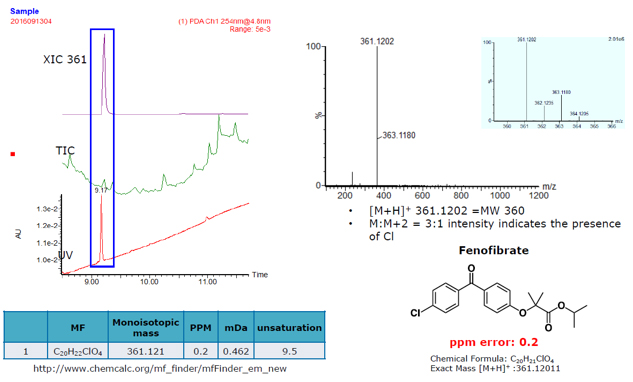
A próxima etapa foi um estudo de spiking (aumento de matriz) que envolveu o “spiking” do API presumivelmente identificado no extrato desconhecido seguido por análise de cromatograma. Uma vez que o tempo de retenção e a massa precisa do composto supostamente identificado e desconhecido foram mostrados para ser o mesmo, a próxima etapa foi construir uma curva padrão usando o composto agora identificado (Figura 5). A área do pico da amostra do desconhecido do extrato (126 mV / s) foi lida contra a curva padrão e a concentração desconhecida determinada. O API desconhecido foi identificado como fenofibrato a 0,14 mg / mL.
SINERGIA DE TECNOLOGIAS ORTOGONAIS
Na Catalent, as tecnologias de RMN e espectrometria de massa são usadas para estudar impurezas ou requalificar materiais. NMR é usado para analisar compostos de moléculas pequenas, bem como peptídeos e proteínas para identificar compostos, confirmar a estrutura dos compostos, avaliar as estruturas secundárias e terciárias das proteínas e determinar a pureza absoluta dos compostos. A espectrometria de massa é usada para caracterização de materiais para garantir identidade, resistência, qualidade, pureza e potência. Juntos, NMR e espectrometria de massa oferecem uma abordagem extremamente poderosa para a caracterização de materiais.
A equipe da Catalent é composta por especialistas altamente qualificados, prontos para aplicar esses métodos para ajudar os clientes a avançar seus projetos de desenvolvimento farmacêutico.
Nenhum comentário:
Postar um comentário