Por Bilel Khedir, Opalia Pharma Recordati Group

Desenvolver um medicamento com múltiplas dosagens tem várias vantagens: ajuda a atender às necessidades individuais dos pacientes, oferece uma vantagem para medicamentos que requerem titulação, evita a quebra de comprimidos (o que é ruim para a estabilidade do medicamento) 1 e, no caso de medicamentos genéricos , contribui para melhorar o acesso a medicamentos de qualidade em todo o mundo. 2 No entanto, para um medicamento genérico, testes em humanos geralmente são necessários para demonstrar a equivalência, mas isenções podem ser consideradas em três casos principais: bioequivalência auto-evidente, bioisenções baseadas no sistema de classificação biofarmacêutica (BCS) e bioisenções de força. 2
Uma bioisenção de força geralmente é reivindicada para uma dosagem adicional quando a bioequivalência foi estabelecida para uma dosagem e a bioisenção BCS não é aplicável. 2 Seis fatores precisam ser considerados com bioisenções de força adicional: 2
- A farmacocinética da substância medicamentosa
- A semelhança do processo de fabricação
- A composição qualitativa
- A composição quantitativa
- O perfil de dissolução
- A metodologia usada para analisar os perfis de dissolução
A mesma formulação poderia obter uma bioisenção de dosagem nos EUA, mas não na UE (e vice-versa) devido a diferenças nos requisitos de formulação. O mesmo conjunto de dados de estudos de dissolução pode produzir perfis semelhantes (portanto, medicamentos semelhantes) nos EUA, mas não semelhantes de acordo com os padrões da UE (e vice-versa) devido a diferenças na escolha do último ponto de tempo. Neste artigo, discutirei os requisitos da EMA e da FDA e os desafios associados ao reivindicar uma bioisenção de força.
1. Farmacocinética da Substância Medicamento
A força na qual os estudos de bioequivalência são feitos é determinada pela farmacocinética (PK) da substância medicamentosa. A definição de farmacocinética linear e a força para escolher se a PK é linear ou não linear diferem entre as perspectivas da EMA e da FDA.
EMA
A farmacocinética linear é definida como uma relação dose-ajustada da área sob a curva (AUC) das diferentes dosagens dentro de ± 25% (75%–133%). 2 A diretriz da EMA sobre bioequivalência define a farmacocinética linear como ocorrendo quando a diferença na AUC média ajustada à dose não é superior a 25% ao comparar a dosagem estudada (usada no estudo de bioequivalência) e a dosagem para a qual uma bioisenção é considerada. 3
Farmacocinética linear: Em geral, os estudos de BE são feitos na dosagem mais alta, mas a EMA aceita a dosagem mais baixa em dois casos:
- A substância medicamentosa é altamente solúvel.
- A dosagem mais alta não pode ser administrada por motivos de segurança/tolerabilidade.
Se ocorrerem problemas na sensibilidade do método analítico, a administração de uma dose maior é tolerada. 3
Farmacocinética não linear: Existem duas possibilidades:
- Aumento mais do que proporcional na AUC com o aumento da dose dentro do intervalo de dose terapêutica: A bioequivalência deve ser estabelecida utilizando apenas a dosagem mais elevada.
- Aumento menos do que proporcional na AUC com o aumento da dose acima do intervalo terapêutico: Neste caso, são necessários dois estudos de bioequivalência com as dosagens mais altas e mais baixas.
Exceção: Se a não linearidade for causada por saturação de absorção e solubilidade não limitada (os produtos genéricos e de marca não contêm excipientes que possam alterar a motilidade gastrointestinal ou proteínas de transporte e o restante das condições para a bioisenção de dosagem for atendida), é suficiente para demonstrar a bioequivalência com a força mais baixa ou uma força na faixa linear. 3
FDA
A FDA não fornece uma definição de farmacocinética linear semelhante à dada pela EMA. 1
Farmacocinética linear: A FDA recomenda conduzir o estudo BE na dosagem mais alta. O uso de menor resistência é permitido por razões de segurança. 4
Farmacocinética não linear: Se a farmacocinética não for linear, a questão da aceitabilidade de uma bioisenção de dosagem é uma questão de revisão da agência, a menos que seja publicada uma orientação específica do produto que contenha detalhes. 4
2. Similaridade do processo de fabricação
A EMA e a FDA exigem o mesmo processo de fabricação para todos os pontos fortes. Ter o mesmo processo de fabricação significa que ambos os medicamentos (de baixa e alta dosagem) devem ter a mesma forma farmacêutica (se uma dosagem for um comprimido, a outra deve ser um comprimido, não uma cápsula e vice-versa). Mas, ao contrário do FDA, o EMA aceita diferentes locais de fabricação para os pontos fortes do medicamento em questão. 2
3. Composição Qualitativa
A EMA e a FDA exigem que todos os ingredientes ativos e inativos sejam iguais. No entanto, sabores, cores, revestimentos (exceto mudanças entre filmes e revestimentos de açúcar) e tintas podem ser diferentes. 2
4. Composição Quantitativa
FDA
A FDA espera que o núcleo de cada força esteja na mesma proporção. Por outro lado, para substâncias medicamentosas de alta potência/baixa porcentagem ( ≤10%) 2 , nenhuma proporcionalidade é exigida, mas três condições devem ser atendidas:
- O peso total da forma de dosagem permanece o mesmo para todas as dosagens (±10% tolerado).
- Os mesmos ingredientes inativos são usados.
- A mudança em qualquer força é obtida alterando a quantidade do ingrediente ativo e um ou mais ingredientes inativos .
A FDA afirma que outras diferenças podem ser aceitas com a devida justificativa. 1
A FDA também espera que cada força tenha a mesma forma. 2
EMA 1
Para o EMA, a composição quantitativa deve estar na mesma proporção; desvio desta regra é aceito sob três condições:
- A quantidade da substância ativa é inferior a 5% do peso do núcleo do comprimido ou do conteúdo da cápsula.
- As quantidades dos diferentes excipientes principais são as mesmas para as dosagens em questão.
- A quantidade de um enchimento é alterada para contabilizar a alteração da substância ativa.
5. Estudos de dissolução in vitro
Perfis comparativos de dissolução in vitro são exigidos pela FDA e EMA, mas existem algumas diferenças em termos de detalhes sobre: 2
- Os produtos a serem comparados
- O número de unidades
- O aparelho necessário
- A mídia necessária
FDA 2
Produtos a serem comparados: Diferentes pontos fortes do produto de teste e do produto comparador (marca).
Número de unidades: 12
Aparelho necessário: Paddle (50 rpm) e cesto (100 rpm) são usados.
Mídia necessária: A FDA exige testes no meio de controle de qualidade acordado. O uso de surfcatnat é aceito.
Apresentação dos dados de dissolução: Um conjunto completo de perfis de dissolução
Linha de produtos de teste (genérica) :
- Dosagem de referência (biolote: o lote usado no estudo de bioequivalência) (por exemplo, a dosagem de 100 mg de medicamento genérico)
- Força de teste (por exemplo, 50 mg de medicamento genérico)
Linha de produtos de referência (marca) :
- Duas dosagens (potência de 100 mg do medicamento de marca e concentração de 50 mg do medicamento de marca)
- A comparação estatística só é necessária dentro da linha de produtos de teste, tendo como referência o biolote da dosagem de referência.
EMA 2
Produtos a serem comparados: A EMA exige que estudos de dissolução sejam feitos dentro da linha de produtos de teste.
Número de unidades: 12
Aparelho necessário: Paddle (50 rpm) e cesto (100 rpm) são usados.
Meio necessário: Meio de controle de qualidade e três pHs diferentes na faixa de pH fisiológico: ácido clorídrico 0,1 N (pH 1,2), tampão acetato (pH 4,5) e tampão fosfato (pH 6,8). O uso de surfactante só é possível no meio QC.
Apresentação dos dados de dissolução:
Linha de produtos de teste (genérica):
- Dosagem de referência (biolote: o lote usado no estudo de bioequivalência) (por exemplo, a dosagem de 100 mg de medicamento genérico)
- Força de teste (por exemplo, 50 mg de medicamento genérico)
- A comparação estatística é necessária entre as duas forças da linha de produtos de teste em cada meio testado.
Linha de produtos de referência (marca):
- Os dados de dissolução das dosagens do produto de referência são necessários apenas em alguns casos, que são discutidos na próxima seção.
6. A Metodologia Utilizada para Analisar os Perfis de Dissolução
Os perfis de dissolução devem ser comparados usando f2, cálculo do fator de similaridade, a menos que a dissolução dos medicamentos seja muito rápida. Para a FDA e a EMA, a dissolução muito rápida ocorre quando mais de 85% da droga é dissolvida tanto para o produto de referência quanto para o produto de teste em 15 minutos. 5
O fator de similaridade f2 é um parâmetro estatístico usado para demonstrar similaridade entre perfis de dissolução. Proposto em 1996 por Moore e Flanner, f2 pode ser descrito como a transformação recíproca da raiz quadrada da soma do erro quadrado e é uma medida da similaridade na dissolução percentual entre dois perfis: 5
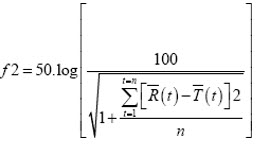
Análise Estatística 5
Para obter resultados confiáveis, ambas as agências (EMA e FDA) impõem certas condições ao calcular o fator de similaridade que cobrem três aspectos principais:
- Número mínimo de pontos de tempo
Para comparação do perfil de dissolução, a EMA e a FDA exigem um mínimo de três pontos de tempo. A amostragem deve ser feita pelo menos a cada 15 minutos de acordo com ambas as agências, mas a amostragem em intervalos de 5 e 10 minutos pode ser útil para a dissolução rápida de medicamentos e muito útil para caracterizar completamente o perfil de dissolução e evitar obter um f2 tendencioso. Assim, a escolha dos tempos de amostragem apropriados requer um estudo de dissolução preliminar. 5
De acordo com a EMA, o último ponto de tempo a ser incluído na análise f2 é quando qualquer um dos produtos de referência e teste atingir 85% ou uma assíntota for atingida. Por outro lado, o FDA define o último ponto no tempo como o ponto no tempo em que não mais do que um valor médio (produto de teste e referência) é ˃ 85% . 5
Para o mesmo conjunto de dados, a aplicação dessas duas regras pode resultar em um f2 abaixo de 50 e acima de 50. É por isso que a escolha do último ponto no tempo é muito importante em uma análise de f2.
- Coeficiente de critérios de variação
O FDA exige um coeficiente de variação ≤ 20% para os primeiros pontos de tempo (15 minutos ou menos) e ≤ 10% nos outros pontos de tempo. 5
O EMA requer um coeficiente de variação ≤ 20% para o primeiro ponto de tempo e ≤ 10% do segundo ao último ponto de tempo. 5
Interpretação dos resultados dos estudos de dissolução in vitro
FDA : Se o f2 (dentro dos pontos fortes da linha de produto de teste) estiver entre 50 e 100, os perfis de dissolução são considerados semelhantes. No entanto, no caso de bioisenções de força, um f2 de < 50 pode ser devido à falta de condições de afundamento. Isso pode ser demonstrado comparando duas dosagens na mesma dose (linha de produtos de teste). A FDA pode exigir o cálculo f2 da força da linha de produtos da marca caso a caso. 2
EMA : Um f2 entre 50 e 100 para perfis de dissolução também é necessário para provar a similaridade. Se o f2 for menor que 50, isso pode ser devido à falta de condições de afundamento; isso é demonstrado comparando duas dosagens na mesma dose (da linha de produtos de teste). Além disso, a EMA exige um f2 < 50 entre as duas dosagens da linha de produtos da marca para provar que a diferença está relacionada à substância do medicamento e não à formulação. 1
Observe a definição de condições de sumidouro: A Farmacopeia Europeia define as condições de sumidouro como um volume de meio de dissolução equivalente a três a 10 vezes o volume de saturação. De acordo com a US Pharmacopeia, a condição de sumidouro é satisfeita quando o meio de dissolução é capaz de dissolver uma quantidade de fármaco três vezes maior que a quantidade de fármaco a ser testada. 6
Conclusão
Os requisitos para bioisenções de dosagem na UE e nos EUA são diferentes em termos de definição de linearidade farmacocinética, similaridade de composição qualitativa e quantitativa e estudos de dissolução in vitro necessários e sua análise estatística (análise f2). Além disso, a escolha de um produto de referência (produto de marca) é fácil do ponto de vista americano, pois o FDA possui uma lista clara de medicamentos listados de referência (RLD), enquanto os países da UE podem ter diferentes medicamentos de referência.
Essas diferenças podem exigir que as empresas farmacêuticas formulem um medicamento diferente para cada apresentação, o que é muito caro. Assim, um diálogo entre os diferentes órgãos pode ser útil para minimizar essas divergências.
Referências
- Cardot, JM, Garcia-Arieta, A., Paixão, P., Tasevska, I., & Davit, B. (2018). Implementando a bioisenção de força adicional para genéricos: abordagens e desafios recomendados pela EMA para uma submissão ao FDA dos EUA. European Journal of Pharmaceutical Sciences, 111, 399-408.
- Crane, C., Santos, GML, Fernandes, EAF, Simon, C., Tam, A., Triana, DG, ... & Garcia-Arieta, A. (2019). Os Requisitos para Bioisenções de Força Adicional para Formas de Dosagem Oral Sólida de Liberação Imediata em Reguladores e Organizações Participantes do Programa Internacional de Reguladores Farmacêuticos: Diferenças e Comunalidades. Journal of Pharmacy & Pharmaceutical Sciences, 22, 486-500.
- Agência Europeia de Medicamentos (EMA). (2010) Diretriz sobre a investigação da bioequivalência https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf
- Food and Drug Administration (FDA). (2021) Estudos de bioequivalência com parâmetros farmacocinéticos para medicamentos enviados sob uma orientação preliminar da ANDA https://www.fda.gov/media/87219/download
- Diaz, DA, Colgan, ST, Langer, CS, Bandi, NT, Likar, MD e Van Alstine, L. (2016). Requisitos de similaridade de dissolução: quão semelhantes ou diferentes são as expectativas regulatórias globais? A revista AAPS, 18, 15-22.
Sobre o autor:
 Bilel Khedir trabalha para o Opalia Pharma Recordati Group como gerente de projeto de desenvolvimento farmacêutico e especialista em garantia de qualidade. Ele também é farmacêutico e estudante de mestrado em desenvolvimento de medicamentos. Sua experiência inclui conduzir estudos de pré-formulação e formulação de medicamentos, validação de métodos analíticos, estudos de bioequivalência in vitro e redigir dossiês de autorização de comercialização de novos medicamentos (formato CTD). Em sua função anterior na Opalia como farmacêutico de garantia de qualidade, ele supervisionou a validação da limpeza, incluindo atividades de limpeza e desinfecção de rotina, e executou estratégias de melhoria contínua. Você pode contatá-lo em khedir.b@opaliarecordati.com .
Bilel Khedir trabalha para o Opalia Pharma Recordati Group como gerente de projeto de desenvolvimento farmacêutico e especialista em garantia de qualidade. Ele também é farmacêutico e estudante de mestrado em desenvolvimento de medicamentos. Sua experiência inclui conduzir estudos de pré-formulação e formulação de medicamentos, validação de métodos analíticos, estudos de bioequivalência in vitro e redigir dossiês de autorização de comercialização de novos medicamentos (formato CTD). Em sua função anterior na Opalia como farmacêutico de garantia de qualidade, ele supervisionou a validação da limpeza, incluindo atividades de limpeza e desinfecção de rotina, e executou estratégias de melhoria contínua. Você pode contatá-lo em khedir.b@opaliarecordati.com .





 Vias de medicação parenteral - fonte Basicmedical Key
Vias de medicação parenteral - fonte Basicmedical Key Fonte: Excipientes parenterais sépticos
Fonte: Excipientes parenterais sépticos
