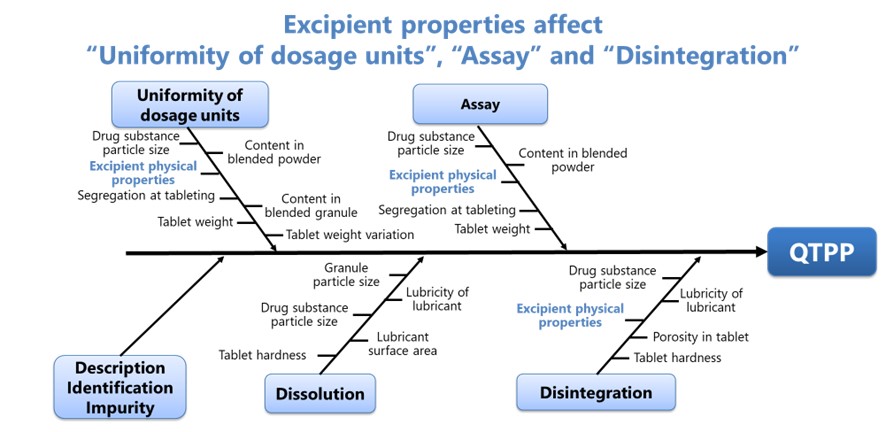
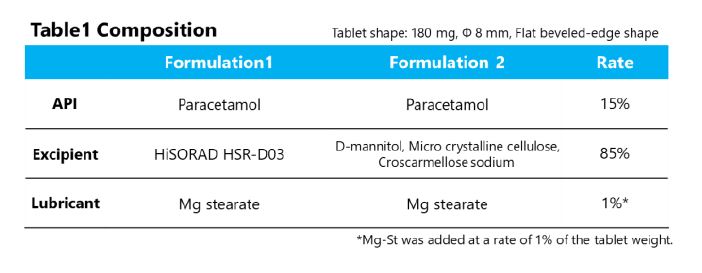
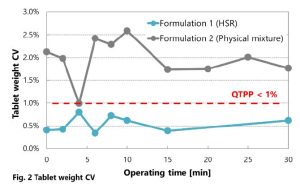
Escolhendo um fabricante de secagem por pulverização: por que a flexibilidade e a personalização são importantes
Por Ana Vilão, Gerente de Spray Drying, Esteve Química

A secagem por pulverização de ingredientes farmacêuticos ativos (APIs) é uma tecnologia amplamente utilizada para aumentar a biodisponibilidade de materiais pouco solúveis, secar produtos farmacêuticos sensíveis ao calor e realizar engenharia de partículas. A secagem por pulverização é um processo no qual uma solução de API e um polímero são rapidamente secos para produzir um pó amorfo. O facto de permitir a personalização das características do pó, nomeadamente a solubilidade, torna-o uma tecnologia preferencial para o número crescente de moléculas que se tornaram candidatas farmacêuticas nos últimos anos.
Processo de Secagem por Pulverização
A secagem por pulverização é um processo de secagem contínua no qual uma solução líquida preparada é seca em um pó sólido amorfo. Envolve a atomização do líquido em gotículas muito pequenas (pluma de spray) usando um atomizador ou bico de spray. Existem vários tipos de atomizadores que podem ser usados (dois fluidos, de pressão, ultrassônicos ou rotativos) dependendo do processo. Na indústria farmacêutica, os mais comuns são bico de dois fluidos e bico de pressão. O processo de atomização ocorre dentro da câmara de secagem, onde as pequenas gotas interagem diretamente com uma corrente de gás quente de secagem que irá evaporar o solvente e permitir a formação da partícula. Existem algumas variações na configuração do gás de secagem. Pode ser co-corrente ou contra-corrente. Os sistemas também podem ter recirculação ou não, conhecidos como sistemas de malha fechada e malha aberta, respectivamente. Os secadores por pulverização mais comumente usados na secagem de ingredientes farmacêuticos ativos operam em co-corrente e em circuito fechado para fins de otimização do processo. Ambos os sistemas de solventes aquosos e orgânicos podem ser usados no processo de secagem por pulverização de pós farmacêuticos, e diferentes tipos de gás de secagem podem ser utilizados, como ar, nitrogênio ou argônio.
A formação da partícula ocorre por meio de secagem rápida dentro da câmara de secagem, após o que as partículas são então separadas da corrente de gás por meio de um ciclone e/ou filtro de mangas. Os ciclones são a opção preferencial utilizada nos processos de secagem por pulverização farmacêutica, pois podem ser projetados para alta eficiência de recuperação e consistem em um mecanismo simples, sem partes móveis, onde o fluxo de gás contendo as partículas é alimentado tangencialmente no topo do ciclone. Um vórtice é criado dentro do ciclone e o produto cairá no fundo do equipamento onde é coletado.
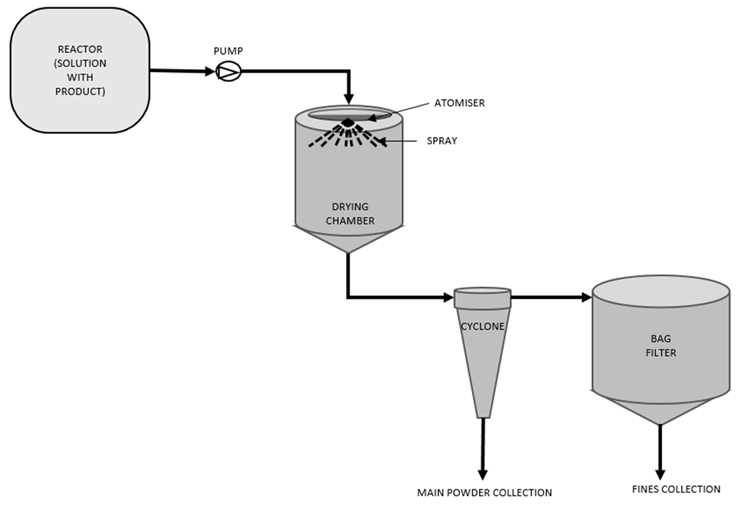
A secagem por pulverização é uma das tecnologias utilizadas para a engenharia de partículas na indústria farmacêutica. Ao projetar ou projetar as partículas, os atributos finais do pó podem ser manipulados e controlados. O processo de secagem por pulverização permite o controle de propriedades como a morfologia da partícula, o tamanho da partícula, a densidade do pó e o nível de umidade ou solvente residual no pó. Essas características podem ser projetadas em grande medida para atingir as metas exigidas de um processo a jusante. A secagem por pulverização normalmente produz partículas amorfas quase esféricas com características de fluxo favoráveis.
Spray Drying na Esteve Química (EQ)
Melhorar a eficiência e a confiabilidade dos processos diante de prazos apertados é uma das principais especialidades da Esteve Química. Ela opera como uma organização de fabricação por contrato (CMO) boutique, oferecendo aos clientes a flexibilidade de ditar suas necessidades individuais dentro da estrutura de qualidade e serviço da Esteve Química.
A Esteve Química também trabalha para ser flexível diante dos desafios do processo, para que um problema no projeto não se traduza automaticamente em atraso na fabricação. Por mais de uma década, a Esteve Química estabeleceu uma equipe central para cada um de seus projetos, projetada para manter um contato constante entre o cliente e as contrapartes da Esteve Química. Essas equipes principais, lideradas por um gerente de projeto e envolvendo especialistas em controle de qualidade, P&D, produção, aquisição, EHS e garantia de qualidade, gerenciam os aspectos técnicos do projeto e mantêm uma comunicação fluente entre si e com suas contrapartes durante a vida de um projeto.
Escolher o CMO certo para um processo requer uma avaliação abrangente dos pontos fortes de um parceiro em potencial – desde as capacidades de uma organização até seu histórico, suas instalações e experiência. Encontrar o parceiro certo pode ser uma tarefa complexa. Isso é particularmente verdadeiro para empresas que buscam terapêuticas com químicas complexas ou necessidades de secagem por pulverização. Encontrar um CMO com experiência e flexibilidade para permitir retornos precisos, impulsionar a otimização contínua e promover a segurança e a eficácia é essencial para proteger a expansão dessas terapias.
A secagem por pulverização para intermediários farmacêuticos é uma das áreas de especialização bem estabelecidas da empresa. A Esteve Química possui experiência significativa em secagem por pulverização e seus sistemas e processos de qualidade foram aprovados por órgãos reguladores em todo o mundo, incluindo o FDA dos EUA e agências regionais. Nosso prazo médio para uma aplicação de secagem por pulverização, desde a transferência de tecnologia até a conclusão de uma campanha de avaliação, é de seis meses, e nossa equipe experiente pode oferecer soluções flexíveis e personalizadas para atender aos requisitos do processo de secagem por pulverização.
Equipamentos e capacidades
Localizada dentro do perímetro da fábrica de Celrà (Espanha), a planta de secagem por pulverização está se posicionando como um forte CMO para atender empresas em todo o mundo. Equipada com instalações cGMP de última geração e laboratórios de caracterização de formas sólidas, a Esteve Química oferece serviços integrados de P&D, analíticos e de fabricação que podem apoiar um processo desde o desenvolvimento inicial até o aumento de escala e a transferência de tecnologia.
A equipe qualificada de cientistas e engenheiros da Esteve Química trabalha em colaboração com o cliente para atender às suas necessidades. Triagem de solventes e polímeros, desenvolvimento de design de qualidade, engenharia de partículas e otimização de processos são alguns de seus serviços mais solicitados.

Encontrar um fabricante de secagem por spray flexível e sob medida
Qualquer empresa farmacêutica que contempla uma estratégia de terceirização significa que está pensando em confiar a uma organização externa a fabricação de seu produto. Os produtos farmacêuticos passam por anos de desenvolvimento e investimentos significativos que pesam muito nas organizações na hora de escolher um fornecedor externo. Portanto, esta decisão requer uma abordagem concertada para avaliar cuidadosamente os potenciais parceiros.
Encontrar um CMO disposto a trabalhar de perto com seus clientes para entender seus processos, produtos e objetivos é fundamental para o sucesso comercial. Igualmente importante é a flexibilidade de um CMO diante de questões emergentes – eles são capazes de se adaptar a possíveis contratempos? Eles priorizarão o sucesso de um projeto em face de prioridades concorrentes? Avaliar um CMO com base em seus intangíveis, incluindo suas atitudes em relação à ética de trabalho, cronogramas e expectativas, pode ser tão ou mais importante do que seus equipamentos ou instalações.
Na Esteve Química, esse compromisso com o sucesso do cliente permeia todos os segmentos de negócios. Esse compromisso se reflete na maneira como adapta sua abordagem a projetos individuais. Onde muitos CMOs grandes ou médios trabalharam para cultivar uma abordagem de tamanho único que serve para cobrir suas bases e aplicar amplamente em seus portfólios de projetos, a Esteve Química trabalha para incorporar os próprios fluxos de trabalho do cliente em sua abordagem. É uma estratégia que funciona para todos os tipos de empresas farmacêuticas, seja você uma grande farmacêutica, biotecnologia de médio porte ou até mesmo uma startup menor, você pode esperar um alinhamento individual entre os membros de sua própria equipe e a equipe da Esteve Química .
Com uma cadeia de suprimentos robusta e uma vasta experiência técnica, a Esteve Química pode oferecer aos clientes confiabilidade e flexibilidade incomparáveis em uma variedade de indicações de doenças e modalidades terapêuticas. Com sua abordagem colaborativa para o gerenciamento de projetos, juntamente com um histórico comprovado de transferência e dimensionamento de processos de forma rápida e contínua, a Esteve Química possui pessoal e recursos para ajudar os parceiros a alcançar um sucesso comercial duradouro. Para saber mais sobre secagem por spray, clique aqui , ou visite esteve.com .
Sobre o autor
 Ana Vilão é licenciada em Engenharia Química pelo Instituto Superior Técnico (Portugal) e Chartered Engineer (MIChemE). Ela tem mais de 15 anos de experiência na indústria na Esteve Química, Thermo Fisher Scientific, GSK, Pfizer e Hovione. Ela é Gerente de Secagem por Spray da Esteve Química e, nesta posição, é responsável pelas Operações Farmacêuticas de Medicamentos Intermediários (DPI), que inclui uma Planta de Secagem por Spray de última geração. Ela lidera uma equipe de operadores, engenheiros e cientistas responsáveis pelo desenvolvimento, ampliação e fabricação de processos DPI. Ingressou na Esteve Química em 2020 como Industrial Process Lead e no ano seguinte assumiu a gestão do Departamento de Spray Drying.
Ana Vilão é licenciada em Engenharia Química pelo Instituto Superior Técnico (Portugal) e Chartered Engineer (MIChemE). Ela tem mais de 15 anos de experiência na indústria na Esteve Química, Thermo Fisher Scientific, GSK, Pfizer e Hovione. Ela é Gerente de Secagem por Spray da Esteve Química e, nesta posição, é responsável pelas Operações Farmacêuticas de Medicamentos Intermediários (DPI), que inclui uma Planta de Secagem por Spray de última geração. Ela lidera uma equipe de operadores, engenheiros e cientistas responsáveis pelo desenvolvimento, ampliação e fabricação de processos DPI. Ingressou na Esteve Química em 2020 como Industrial Process Lead e no ano seguinte assumiu a gestão do Departamento de Spray Drying.