Lista de Verificação de Desenvolvimento de Produto: Considerações para Cada Estágio do Processo de Desenvolvimento de Medicamentos

Desenvolver um novo produto biofarmacêutico é uma jornada longa e de alto risco. Leva, em média, pelo menos 10 anos e mais de US$ 2 bilhões para trazer com sucesso um novo medicamento ao mercado, e apenas 10 a 15 por cento dos produtos recebem aprovação regulatória.[1]
Um plano abrangente e o conhecimento regulatório e terapêutico correto podem acelerar significativamente o cronograma de desenvolvimento e aumentar a probabilidade de sucesso do marketing, especialmente para pequenas empresas de biotecnologia e farmacêuticas especializadas que trabalham com tempo e recursos limitados. Este post fornece uma visão geral das atividades críticas de planejamento e marcos no processo de desenvolvimento de medicamentos e descreve importantes considerações regulatórias em cada estágio.
Etapa 1: Planejamento
O planejamento é a base do sucesso e envolve uma longa lista de verificação de tarefas obrigatórias. O planejamento perfeito não é crítico, mas a falta de planejamento certamente prejudicará seus esforços. Com um plano estabelecido desde o início, você pode fazer ajustes em cada fase do programa, conforme necessário.
Para otimizar um programa de desenvolvimento de medicamentos para uma nova entidade terapêutica, pense cuidadosamente nas seguintes etapas:
- Estabeleça as metas do programa
- Defina a estratégia regulatória
- Esclarecer os requisitos críticos para aprovação
- Delinear o plano de desenvolvimento do produto no início
O plano de desenvolvimento de medicamentos deve incluir informações críticas sobre o produto como:
- Uma indicação e declaração de uso descrevendo a doença ou condição alvo
- Uma definição de uso pretendido: para tratamento, prevenção, mitigação, cura, alívio ou diagnóstico
- Forma de dosagem e via de entrega
- Expectativas de eficácia, medidas por resultados ou desfechos definidos
- Expectativas de segurança
Também essencial na fase de planejamento é determinar o escopo geográfico de comercialização e as diretrizes regulatórias aplicáveis. Revise minuciosamente essas considerações comerciais desde o início para o planejamento estratégico de mercado:
- Os produtos competitivos já estão disponíveis?
- Como o novo medicamento é diferente das opções de tratamento existentes?
- Qual é a participação de mercado potencial para o medicamento em investigação?
- O produto pode se qualificar para uma designação especial pela Food and Drug Administration ou outro órgão regulador com base em seu potencial para atender a necessidades médicas não atendidas ou uma condição para a qual os tratamentos não existem ou são inadequados?
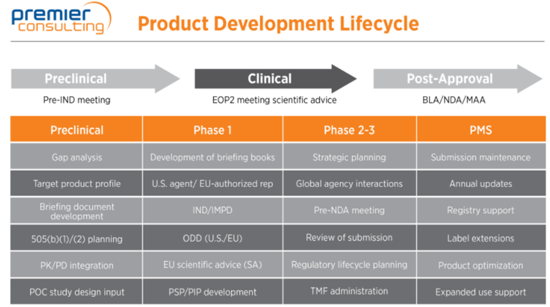
Estágio 2: Desenvolvimento pré-clínico
O primeiro passo na avaliação de uma nova entidade química ou molecular é a realização de estudos pré-clínicos in vitro e depois in vivo que definem as características de segurança relevantes.
Estudos de farmacologia de segurança avaliam como a droga afeta os sistemas respiratório, cardiovascular e nervoso central, entre outros.
Os estudos farmacodinâmicos estabelecem o mecanismo de ação da droga e contribuem para a seleção da dose para estudos clínicos.
Os estudos de toxicologia definem quanto do medicamento pode ser administrado com segurança e com que frequência, incluindo:
- A dose máxima tolerada
- O nível de efeito adverso não observado, que determina a primeira dosagem em humanos
- Se as doses repetidas são tóxicas - por exemplo, com uso crônico no tratamento de diabetes ou hipercolesterolemia
Os ensaios farmacocinéticos caracterizam como o fármaco é absorvido, distribuído, metabolizado e excretado.
Testes adicionais para carcinogenicidade, imunotoxicidade, interações medicamentosas e outros efeitos continuam durante o desenvolvimento clínico.
A química, a fabricação e o planejamento de controles na fase pré-clínica garantem que a fabricação do medicamento seja cuidadosamente considerada em todas as etapas, desde os lotes de desenvolvimento até a apresentação comercial. Isso inclui:
- Definição de ingredientes ativos e matérias-primas
- Desenvolvimento e ampliação do processo de fabricação
- Configurando controles em processo
- Desenvolvimento e qualificação de métodos analíticos
- Determinando os requisitos de estabilidade
- Estabelecer orientação sobre como definir e justificar as especificações do produto
O desenho do ensaio clínico é a etapa final da fase pré-clínica. Os cientistas determinam a quantidade apropriada do medicamento para obter segurança e eficácia por meio de ensaios de microdose, dose única ascendente ou múltiplas doses ascendentes. Para ensaios randomizados, as decisões devem ser feitas sobre o tipo de cegamento. A nova entidade será testada contra um comparador aprovado ou placebo? Estudos de segurança suplementares também podem ser necessários em uma ou mais fases.
Quando o desenho do estudo for finalizado, um novo medicamento sob investigação ou pedido de ensaio clínico análogo deve ser apresentado à FDA, à Agência Europeia de Medicamentos e/ou aos órgãos reguladores de quaisquer outros países onde você planeja realizar o estudo. Este pedido solicita autorização para administrar o biofármaco a humanos. Solicitar uma reunião pré-IND com a agência antes do envio pode ser útil para resolver dúvidas, abordar preocupações e evitar surpresas logo antes do início do julgamento. A FDA exige um pacote de informações com materiais e dados relevantes pelo menos um mês antes de uma reunião solicitada.
Estágio 3: Desenvolvimento clínico
Nos estágios iniciais do desenvolvimento clínico, o foco da investigação é o monitoramento da segurança.
Os estudos de fase 1 geralmente avaliam a segurança em um pequeno número de voluntários saudáveis. Avaliações preliminares de eficácia em pacientes afetados podem ser incluídas na Fase 1 se a terapia estiver sendo avaliada em pacientes e se destinar a atender uma necessidade médica não atendida ou com risco de vida.
Os ensaios de fase 2 visam determinar se o medicamento é seguro e eficaz em uma coorte maior de pacientes com a condição em tratamento. Após esse estágio, uma reunião de final da Fase 2 com o FDA deve ser agendada para revisar o desenho do estudo principal (Fase 3). O objetivo do ensaio clínico principal é demonstrar que o medicamento investigado tem melhor eficácia do que o padrão de tratamento estabelecido.
Os ensaios de fase 3 avaliam se o medicamento é seguro e eficaz em uma população maior de pacientes. Se os dados demonstrarem isso, o próximo passo é preparar um pedido de marketing – um pedido de novo medicamento ou um pedido de licença de produtos biológicos – para envio ao FDA. Semelhante ao IND, uma reunião pré-NDA/BLA com a agência é útil para esclarecer as expectativas e discutir os requisitos de apresentação e formato do pedido.
Etapa 4: monitoramento de segurança pós-comercialização
Após a aprovação de comercialização, a FDA exige monitoramento de segurança pós-comercialização assim que os produtos forem disponibilizados ao público. O planejamento de farmacovigilância deve delinear as ações a serem tomadas quando ou se os eventos adversos aumentarem com a absorção do medicamento por um número maior de pacientes. Ensaios de fase 4 podem ser necessários para estudar mais as características do medicamento em relação à segurança, eficácia, indicações expandidas e novas formulações ou vias de administração. Essas mudanças podem resultar em exclusividade de mercado estendida sob proteção de patente adicional. Os inovadores também precisam antecipar e planejar mudanças regulatórias em andamento, como atualizações de rotulagem.
A Premier Consulting ajuda os patrocinadores a obter um desenvolvimento mais rápido e uso mais eficiente de recursos limitados, integrando estratégias e operações pré-clínicas e clínicas. Nossos serviços de consultoria fornecem acesso a uma rede global de especialistas em desenvolvimento de produtos e regulamentação para ajudar pequenas empresas de biotecnologia e farmacêuticas especializadas a projetar e executar planos abrangentes para o desenvolvimento de produtos, desde a descoberta inicial até o IND e o gerenciamento do ciclo de vida pós-aprovação. Isso permite que os patrocinadores mantenham o desenvolvimento de produtos avançando com eficiência, reduzindo o risco e maximizando a oportunidade de sucesso comercial. Entre em contato para agendar uma reunião com nossa equipe.
Fontes:
[1] PhRMA. Pesquisa e Desenvolvimento Biofarmacêutico: O Processo por Trás de Novos Medicamentos, 2015.
Nenhum comentário:
Postar um comentário