O poder discriminativo dos métodos de dissolução nos EUA e na Europa
Por Bilel Khedir, Opalia Pharma Recordati Group

Os métodos de dissolução encontrados no banco de dados de dissolução da FDA, nas monografias da US Pharmacopeia (USP) para produtos acabados ou em qualquer outro compêndio de monografias para produtos acabados são muito úteis, pois não requerem validação completa antes de serem implementados. Muitos pequenos detalhes podem fazer a diferença ao usar esses métodos. Este artigo discutirá um fator que pode mudar o jogo para testes de dissolução em controle de qualidade ou pesquisa e desenvolvimento de comprimidos de liberação imediata : o poder discriminatório.
O poder discriminatório é determinado pela fabricação de “lotes ruins”, que são lotes fabricados com mudanças significativas nos parâmetros críticos do processo (CPPs) ou atributos críticos do material (CMAs) que podem ter impacto na biodisponibilidade do medicamento. 1 Os lotes ruins também podem ser gerados com alterações quantitativas na formulação. Para ser discriminativo, o método deve detectar os lotes ruins quando testado. 1 Assim, um método de dissolução com bom poder discriminatório fornece uma visão mais ampla sobre a qualidade e o desempenho do medicamento. A não verificação do poder discriminatório de um método de dissolução, incluindo métodos compendiais, pode ter consequências perigosas no controle de qualidade e no desenvolvimento de medicamentos.
Poder discriminatório dos métodos de dissolução no controle de qualidade
De acordo com a USP, o teste de dissolução deve, na maioria dos casos, ser discriminatório para os atributos críticos de qualidade do produto. 2 A Agência Europeia de Medicamentos (EMA) afirma em seu documento de reflexão sobre a especificação de dissolução para produtos genéricos sólidos orais de liberação imediata com ação sistêmica que, idealmente, todos os não bioequivalentes devem ser detectados pelos resultados do teste de dissolução in vitro. 1 Assim, os testes de dissolução no controle de qualidade (QC) são feitos para garantir a consistência de lote para lote, detectando alterações em CPPs e CMAs e, assim, garantindo um desempenho in vivo aceitável. 3
Se um método de dissolução não for discriminatório, alterações significativas em CPPs e CMAs não seriam detectadas e os lotes não bioequivalentes passariam facilmente no teste.
O grande problema aqui é que muita gente da indústria farmacêutica acredita que os métodos de dissolução listados nas monografias da USP são aplicáveis sem verificar seu poder discriminatório. Na realidade, esses métodos precisam ser testados quanto ao seu poder discriminatório antes do uso. Por exemplo, uma avaliação do poder discriminatório do método de dissolução usado para comprimidos contendo candesartan cilexetil foi realizada em produtos genéricos no Egito, e os autores descobriram que o método não era discriminativo. 4
A Perspectiva Americana: Farmacopeia dos EUA e FDA
Para fabricantes de medicamentos genéricos, o uso de métodos de dissolução da USP ou mesmo métodos listados no banco de dados de dissolução da FDA não é possível sem a verificação do poder discriminatório. 5
Também é importante observar que os critérios de aceitação definidos nas monografias da USP não são “tamanho único” e devem ser determinados para o seu produto específico, considerando os perfis de liberação durante o desenvolvimento do produto. O valor Q e o tempo de amostragem são selecionados com base no poder discriminatório para atributos críticos de qualidade. 6
Ao desenvolver um método de dissolução para uma forma de dosagem de liberação imediata, a USP recomenda derivar especificações de perfis de dissolução com os seguintes tempos de amostragem: 10 minutos, 15 minutos, 30 minutos, 45 minutos e 60 minutos. Em seguida, o valor Q é selecionado no primeiro ponto de tempo (não menos de 15 minutos) onde pelo menos 85% da quantidade marcada do medicamento é dissolvida. 7
A Perspectiva Europeia: EMA
De acordo com a EMA, a definição do valor Q e o intervalo de tempo que permite a discriminação devem ser derivados do perfil de dissolução do biolote. Isso permitirá a extrapolação dos resultados dos estudos de bioequivalência para os resultados dos lotes comerciais. 1
A escolha do valor Q e do ponto de tempo é muito bem explicada no documento de reflexão da EMA sobre a especificação de dissolução para produtos orais sólidos genéricos de liberação imediata com ação sistêmica . 1
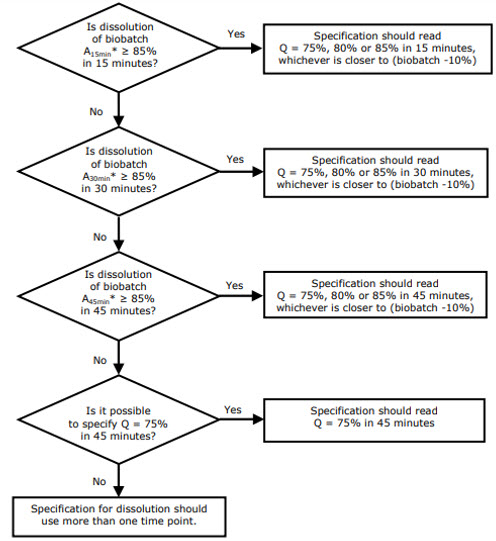
Figura 1: Árvore de decisão dos princípios para definição de especificações com base nos resultados da dissolução do biolote. 1 “A x min” significa “Quantidade (valor médio de 12 unidades) dissolvida em x min”.
Poder discriminatório dos métodos de dissolução em pesquisa e desenvolvimento
Os estudos de dissolução in vitro são muito importantes, pois são usados para selecionar formulações e subsidiar estudos de bioequivalência. Portanto, isso mostra a importância do poder discriminatório dos métodos de dissolução no desenvolvimento de medicamentos genéricos.
Os requisitos da EMA e da FDA em relação ao tipo de estudos de dissolução para bioequivalência são diferentes para comprimidos de liberação imediata. O EMA requer estudos de dissolução em quatro meios: o meio QC e três meios com pH 1,2, pH 4,5 e pH 6,8. 8 Por outro lado, o FDA exige apenas um estudo de dissolução no meio QC com estudos de poder discriminatório enviados (mesmo que o método de dissolução usado seja da USP ou do banco de dados de dissolução do FDA). 9
À primeira vista, os requisitos do FDA podem parecer simples e fáceis de passar (um meio em vez dos quatro meios exigidos pela EMA), mas, na minha opinião, a abordagem americana é baseada em boa ciência e, precisamente, no conceito de poder discriminatório.
Com efeito, se o método de dissolução do CQ for discriminativo “suficiente” e o seu medicamento genérico apresentar um perfil semelhante ao do medicamento de marca, isso significa que o seu medicamento (genérico) é semelhante em termos de qualidade farmacêutica ao produto de marca, e você pode entrar em um ensaio clínico com um bom nível de confiança sem realizar estudos nas outras três mídias exigidas pela EMA.
Supõe-se que os meios adicionais imitem a dissolução in vivo, mas os estudos nesses meios podem ser desafiadores (por exemplo, falta de condições de afundamento, degradação do ingrediente farmacêutico ativo) e podem não refletir o que realmente acontecerá in vivo para uma liberação imediata tábua.
Poder discriminatório de um método de dissolução: exceções
A Perspectiva Americana: USP e FDA
A USP requer um método de dissolução discriminativo na maioria dos casos. 2 Os casos em que o método de dissolução não precisa ser discriminativo estão bem explicados na diretriz da FDA para testes de dissolução e critérios de aceitação para medicamentos em forma de dosagem oral sólida de liberação imediata contendo substâncias medicamentosas de alta solubilidade. De fato, quando o fármaco é altamente solúvel (BCS Classe I ou III) e formulado na forma de liberação imediata, o método de dissolução desenvolvido não precisa ter poder discriminativo. Como consequência, você pode usar o método de dissolução da USP sem verificar o poder discriminatório, ou pode desenvolver um método conforme citado na diretriz e apresentá-lo sem verificar seu poder discriminatório. 10
A Perspectiva Europeia: EMA
Para a EMA, as substâncias medicamentosas altamente solúveis (BCS Classe I ou III) dificultam muito a detecção de alterações na formulação ao realizar a verificação do poder discriminatório. Como consequência, o método de dissolução deve ser usado sem verificação adicional ou substituído por um teste de desintegração, pois o teste de desintegração seria mais discriminativo neste caso. 1
Acho que substituir o teste de dissolução por um teste de desintegração neste caso é muito útil e cientificamente sólido, mas seria muito controverso, pois a Farmacopeia Européia na monografia geral de comprimidos aceita a remoção de testes de desintegração quando testes de dissolução são feitos para comprimidos de liberação imediata.
Conclusão
O poder discriminatório é um fator muitas vezes negligenciado no desenvolvimento de métodos de dissolução internos e, principalmente, no uso de métodos compendiais de monografias de produtos acabados. Os riscos de usar um método não discriminatório são muito significativos no CQ, pois você pode não conseguir extrapolar os resultados de bioequivalência para seus lotes comerciais e, assim, liberar lotes ruins. Em pesquisa e desenvolvimento, o uso de um método de dissolução não discriminatório pode resultar na seleção de uma formulação que falharia em ensaios clínicos.
As perspectivas européia e americana em relação aos testes de dissolução em CQ e P&D são diferentes, mas ambas enfatizam a importância do poder discriminatório dos métodos de dissolução. Como as especificações de dissolução da USP e da Farmacopeia Europeia não são universais, sua empresa farmacêutica deve desenvolver especificações internas para dissolução e desintegração com base em monografias compendiais (monografias de medicamentos acabados ou monografias gerais) de acordo com os estudos que você fez em seu produto específico.
Referências
- Documento de reflexão sobre a especificação de dissolução para produtos orais sólidos genéricos de liberação imediata com ação sistêmica, EMA/CHMP/CVMP/336031 , 2017 https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper- dissolução-especificação-genérico-sólido-oral-liberação-imediata-produtos-sistêmicos_en.pdf
- USP. Formas farmacêuticas orais - Testes de desempenho <1711> In: USP–NF . Rockville, MD: United States Pharmacopeia. Acessado em 4 de fevereiro de 2023. https://doi.org/10.31003/USPNF_M7083_02_01
- Abend, A., Curran, D., Kuiper, J., Lu, X., Li, H., Hermans, A., ... & Suarez-Sharp, S. (2019). Testes de dissolução no desenvolvimento de medicamentos: relatório resumido do workshop.
- Amer, AM, Allam, AN e Abdallah, OY (2018). Avaliação do poder discriminatório do método de dissolução USP para comprimidos de candesartan cilexetil por meio de testes de produtos comercializados no Egito. Tecnologia de dissolução , 25 (4), 40-46
- Anand, OM, Yu, LX, Conner, DP e Davit, BM (2011). Teste de dissolução para medicamentos genéricos: uma perspectiva da FDA. A revista AAPS , 13 , 328-335
- Dissolution Technologies (2017), Perguntas e respostas de novembro de 2017, acessado em 28 de janeiro de 2023 [ http://dissolutiontech.com/issues/201711/201711qa.php ]
- USP. O Processo de Dissolução-Desenvolvimento e Validação <1092>. In: USP-NF . Rockville, MD: United States Pharmacopeia. Acessado em 4 de fevereiro de 2023. https://doi.org/10.31003/USPNF_M643_05_01
- European Medicine Agency [EMA].(2010) Diretriz sobre a investigação da bioequivalência https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf
- Food and Drug Administration [FDA]. (2021) Estudos de bioequivalência com parâmetros farmacocinéticos para medicamentos enviados sob uma orientação preliminar da ANDA https://www.fda.gov/media/87219/download
- Food and Drug Administration [FDA]. (2018), Teste de Dissolução e Critérios de Aceitação para Medicamentos em Forma de Dosagem Oral Sólida de Liberação Imediata que Contêm Substâncias Medicamentosas de Alta Solubilidade. https://www.fda.gov/files/drugs/published/Dissolution-Testing-and-Acceptance-Criteria-for-Immediate-Release-Solid-Oral-Dosage-Form-Drug-Products-Containing-High-Solubility- _ Drogas-Substâncias-Orientação-para-Indústria.pdf
Sobre o autor:
 Bilel Khedir trabalha para o Opalia Pharma Recordati Group como gerente de projeto de desenvolvimento farmacêutico e especialista em garantia de qualidade. Ele também é farmacêutico e estudante de mestrado em desenvolvimento de medicamentos. Sua experiência inclui conduzir estudos de pré-formulação e formulação de medicamentos, validação de métodos analíticos, estudos de bioequivalência in vitro e redigir dossiês de autorização de comercialização de novos medicamentos (formato CTD). Em sua função anterior na Opalia como farmacêutico de garantia de qualidade, ele supervisionou a validação da limpeza, incluindo atividades de limpeza e desinfecção de rotina, e executou estratégias de melhoria contínua. Você pode contatá-lo em khedir.b@opaliarecordati.com .
Bilel Khedir trabalha para o Opalia Pharma Recordati Group como gerente de projeto de desenvolvimento farmacêutico e especialista em garantia de qualidade. Ele também é farmacêutico e estudante de mestrado em desenvolvimento de medicamentos. Sua experiência inclui conduzir estudos de pré-formulação e formulação de medicamentos, validação de métodos analíticos, estudos de bioequivalência in vitro e redigir dossiês de autorização de comercialização de novos medicamentos (formato CTD). Em sua função anterior na Opalia como farmacêutico de garantia de qualidade, ele supervisionou a validação da limpeza, incluindo atividades de limpeza e desinfecção de rotina, e executou estratégias de melhoria contínua. Você pode contatá-lo em khedir.b@opaliarecordati.com .
Nenhum comentário:
Postar um comentário