Desenvolvimento de formulações de vacinas: passado, presente e futuro
Diferentes abordagens de formulação de vacina
A situação atual, fortemente influenciada pela pandemia em curso, coloca as vacinas de volta no centro das atenções. Entretanto, as vacinas convencionais e tradicionais apresentam desvantagens, principalmente relacionadas à imunogenicidade, estabilidade e armazenamento do produto final. Freqüentemente, tais produtos requerem a manutenção de uma “rede de frio”, impactando os custos, a disponibilidade e a distribuição das vacinas. Aqui, após relembrar o modo de ação das vacinas e os tipos de vacinas disponíveis atualmente, analisamos o passado, o presente e o futuro da formulação de vacinas. O passado se concentra em formulações convencionais, o presente discute o uso de nanopartículas para aplicação de vacinas e como adjuvantes, enquanto o futuro apresenta adesivos de microagulha como formulação alternativa e via de administração. Finalmente,
Introdução
Os desafiadores meses de 2020 trouxeram à tona as questões críticas associadas à descoberta e formulação de tratamentos eficazes e, em última análise, uma vacina durante epidemias e pandemias. O surgimento de um novo tipo de coronavírus respiratório (síndrome respiratória aguda grave, SARS, CoV ou novo CoV 2019) e seu crescimento na pandemia atual relembram as duas experiências anteriores com CoV, a saber, SARS-CoV e Síndrome Respiratória do Oriente Médio Coronavírus (MERS-CoV). No entanto, o desenvolvimento de vacinas eficazes para SARS-CoV ou MERS-CoV foi retardado ou abandonado assim que a epidemia foi controlada. As análises críticas sobre a preparação para a pandemia após a pandemia de H1N1, destacando a falha em distribuir vacinas suficientes onde eram necessárias, quando eram necessárias, não haviam sido implementadas antes do surgimento do SARS-CoV2.
A atual pandemia destacou também os desafios relacionados à distribuição oportuna de vacinas para a gripe sazonal ou outras doenças, juntamente com a problemática “cadeia de frio”. Esses desafios dependem muito das formulações das vacinas e de suas características (Tabela 1) e, portanto, da pesquisa e das inovações em tecnologia farmacêutica.
Avançando na compreensão do fenômeno de desintegração do tablet - uma atualização sobre estudos recentes
A desintegração é a desagregação de partículas dentro dos comprimidos após a exposição a fluidos aquosos. Sendo uma etapa essencial na cascata de biodisponibilidade, a desintegração é um atributo de qualidade fundamental dos comprimidos de liberação imediata. Embora o fenômeno da desintegração tenha sido estudado por mais de seis décadas, ainda existem algumas lacunas de conhecimento e questões de pesquisa. Três revisões, publicadas em 2015, 2016 e 2017, discutiram a literatura relativa à desintegração do tablet e resumiram a compreensão deste tópico. No entanto, desde então, mais estudos foram publicados, adicionando ao corpo de conhecimento estabelecido. Este artigo dá um passo à frente na compreensão da desintegração, revisando, de forma concisa, as atualizações científicas mais recentes sobre o tema. Inicialmente, revisitamos os mecanismos de desintegração em relação aos três superdesintegrantes mais usados, a saber, amidoglicolato de sódio, croscarmelose sódica e crospovidona. Em seguida, é analisada a influência das condições de formulação, armazenamento, fabricação e meio na desintegração. Isso é seguido por uma digressão sobre novos desintegrantes. Finalmente, destacamos questões de pesquisa não respondidas e imaginamos locais de pesquisa futuros no campo.continue lendo
Como a extrusão pode beneficiar seu produto farmacêutico oral?
O processamento de extrusão tem como objetivo a fabricação contínua de produtos homogêneos e estruturados. Devido à sua imensa adaptabilidade, a extrusão tem sido amplamente utilizada em diversos campos. Está sendo adotado mais recentemente pela indústria farmacêutica em um ritmo sem precedentes para atender a várias necessidades de formulação e fabricação de medicamentos. O processo de extrusão de dupla rosca pode ser usado não apenas para fabricar novos sistemas de distribuição de medicamentos, mas também para substituir os processos de granulação em lote. Durante a extrusão, mistura (s) em pó ou uma mistura granular é compactada, forçada através de um orifício sob condições controladas e finalmente convertida em um produto com forma e densidade definidas. Comparado a um processo de lote tradicional,
Extrusão de Hot Melt
HME evoluiu para uma tecnologia capacitadora
Como um processo de fabricação contínuo, a extrusão foi consolidada na fabricação de produtos farmacêuticos na década de 1980 com o lançamento no mercado do verapamil de liberação sustentada, que contém ingrediente farmacêutico ativo cristalino (API) incorporado em polímeros. Após o lançamento do Verapamil, os pesquisadores se voltaram para a extrusão de fusão a quente (HME) como uma tecnologia capacitadora eficaz para a fabricação de dispersões sólidas amorfas (ASDs) para compostos pouco solúveis. Durante o HME, uma mistura de pó consistindo de API cristalino e polímero (s) é transformada em um extrudado, que contém API disperso molecularmente em uma matriz de polímero. O extrudado pode ser moldado diretamente ou subsequentemente convertido em grânulos ou pelotas para processamento posterior. ASDs projetados racionalmente podem alcançar maior solubilidade aparente e melhor biodisponibilidade.
Por ser um processo contínuo, a extrusão é uma opção econômica e eficiente para reduzir o tempo de produção. Além disso, como o HME é uma tecnologia madura, também é mais escalonável com um controle de processo mais rígido. Em comparação com outras técnicas de fabricação de ADSs, como a secagem por spray, o HME oferece grandes vantagens de pegada menor e processo sem solvente.
A fabricação bem-sucedida de produtos ASD usando HME requer uma compreensão completa das características da formulação, seleção adequada de equipamentos e caracterização abrangente de cada estágio do processo de extrusão. Portanto, é sensato fazer parceria com uma organização de fabricação contratada (CMO) experiente para a entrega de um medicamento consistente e de alta qualidade para entrada no mercado em tempo hábil.
A caracterização completa é essencial para um HME eficaz
Para que o HME atinja seu objetivo pretendido de fabricação de formulações de ASD, a caracterização detalhada de cada aspecto da formulação de ASD é essencial para garantir um processo robusto e qualidade do produto. As propriedades físico-químicas do API, incluindo, mas não se limitando a, solubilidade, temperatura de fusão, estados sólidos, lipofilicidade e estabilidade devem ser completamente caracterizadas. Atenção também deve ser dada à escolha do polímero e suas propriedades. Um polímero ideal deve demonstrar características químicas e físicas adequadas, como propriedades de fluxo, compressibilidade e comportamento termoplástico, incluindo uma temperatura de transição vítrea (Tg) adequada, propriedades reológicas e boa estabilidade térmica. Muitas vezes, um surfactante também é incorporado em uma formulação de ASD para aumentar a carga de droga, aumentar ainda mais a taxa de dissolução, e facilitar o processo de extrusão reduzindo a temperatura do processo. Como a separação de fases pode impactar significativamente a qualidade do produto HME, qualificar a miscibilidade entre todos os componentes na formulação e controlar os níveis de impureza de todos os componentes da formulação é extremamente importante.
A seleção do equipamento e o projeto do processo também podem impactar significativamente a qualidade de um produto HME. Um dos aspectos mais desafiadores no desenvolvimento de um processo de HME para a fabricação de um ASD é atingir o equilíbrio entre a obtenção de um ASD uniforme, fornecendo mistura e dispersão suficientes, enquanto minimiza a degradação da droga e / ou polímero. Um projeto de extrusora de dupla rosca é favorável para aplicações farmacêuticas devido à sua capacidade superior de mistura e menor tempo de residência do material. A extrusão é uma operação unitária integrada que consiste em diferentes zonas funcionais (por exemplo, transporte, mistura, fusão, desgaseificação, modelagem, etc.). A interação entre as propriedades do material e a energia aplicada em diferentes zonas em toda a extrusora é bastante complicada e controles relevantes precisam ser identificados e implementados. Parâmetros de processo individuais,
Quality by Design (QbD) promove uma compreensão completa do produto e do processo de fabricação por meio de uma abordagem sistemática. Fundamentalmente, a qualidade do produto HME é determinada diretamente por parâmetros de sistema chave independentes de escala, incluindo energia específica, distribuição de tempo de residência e pressão. A implantação desses parâmetros de processo independentes de escala é uma maneira ideal de preencher a lacuna entre os atributos de qualidade e os parâmetros independentes de processo. O desenvolvimento do espaço de design em torno dos parâmetros-chave do sistema garante o aumento de escala do processo e flexibilidade de fabricação, mantendo os atributos de qualidade críticos. Além disso, a simulação de extrusão permite um aumento de escala do processo de baixo custo com alta confiança. A simulação de extrusão é particularmente útil quando a similaridade geométrica e as estratégias clássicas de aumento de escala não são aplicáveis.
Granulação Contínua
A granulação é um processo de formação / produção de materiais granulares a partir de substâncias sólidas em pó. O processo de granulação é amplamente utilizado na indústria farmacêutica para melhorar o fluxo, densidade, uniformidade e compressibilidade do material para processamento posterior. Os métodos de granulação podem ser categorizados em granulação úmida, granulação seca e granulação fundida. A seleção de um método apropriado é determinada pela natureza dos materiais de entrada. A granulação por fusão depende da amálgama de um agente de ligação com os materiais de entrada após aquecimento. Em contraste, a granulação úmida usa líquido e um aglutinante para iniciar a formação de agregados. Na granulação a seco, as partículas de pó primárias são normalmente agregadas por compactação e densificação sob alta pressão. A granulação tem sido tradicionalmente um processo em lote. Contudo, a granulação contínua tem recebido cada vez mais atenção, pois oferece vantagens significativas em termos de melhoria da eficiência e redução de custos relacionados ao desenvolvimento, aumento de escala e produção comercial. A AbbVie demonstrou que a granulação contínua por extrusão é uma opção viável para a fabricação de uma variedade de produtos. A granulação por extrusão pode eliminar a operação da unidade de mistura intragranulada alimentando diretamente o ingrediente individual para a extrusora usando alimentadores de perda de peso. Com um projeto de parafuso adequado e configuração de processo, a distribuição unimodal do tamanho de partícula dos grânulos pode ser alcançada sem um processo de moagem, o que pode otimizar significativamente o processo. A mistura eficiente em uma extrusora também permite um produto uniforme com menos solução aglutinante necessária para a granulação úmida, o que subsequentemente reduz o tempo de secagem.
A extrusão oferece muitos benefícios
Amplamente reconhecida por sua capacidade de melhorar a qualidade e as características de processamento de um medicamento, a extrusão é um processo estabelecido para o desenvolvimento de medicamento e fabricação comercial. Se você deseja melhorar a biodisponibilidade, aumentar a solubilidade, mascarar um sabor desagradável, modular a liberação do medicamento ou resolver problemas de estabilidade do medicamento, a extrusão pode ser usada para atingir esses objetivos e muito mais. Para discutir aplicações de extrusão ou para saber mais sobre como AbbVie CMO pode ajudar em seus esforços de desenvolvimento de medicamentos, entre em contato conosco pelo telefone 1-847-938-8524 ou visite www.abbviecontractmfg.com
domingo, 21 de fevereiro de 2021
Aplicação de qualidade por projeto à pesquisa e desenvolvimento farmacêutico
Por Shawn Watson, Chefe de Pesquisa e Desenvolvimento
As origens da qualidade farmacêutica por Design (QbD) podem ser rastreadas até o livro de W. Edwards Deming, Out of Crisis, publicado pela primeira vez em 1982. No livro, Deming apresentou seus quatorze pontos de gestão que tiveram um impacto significativo sobre como as organizações buscam os resultados pretendidos. O terceiro ponto de Deming transformou a forma como alcançamos qualidade; não por meio de testes de pós-fabricação, mas projetando produtos de qualidade desde os estágios iniciais de desenvolvimento.
A orientação do FDA com relação ao QbD começou a tomar forma não muito depois que comecei a trabalhar como químico de bancada, minha primeira função na indústria farmacêutica. O QbD sempre fez parte da minha vida profissional e acredito firmemente que começa nos primeiros estágios do desenvolvimento de medicamentos. Ele também tem sido continuamente refinado pelo FDA em coordenação com a indústria e meu próprio conhecimento tem avançado continuamente com a experiência.
Farmacêutico QbD
O conceito de QbD afirma que a qualidade deve ser projetada em um medicamento com base na compreensão do produto e do processo pelo qual ele é desenvolvido e fabricado. O QbD é frequentemente discutido no contexto de desenvolvimento e fabricação de processos. No entanto, este artigo enfoca como o QbD é aplicado à pesquisa e desenvolvimento (P&D) para gerar melhores resultados em todo o processo de desenvolvimento de medicamentos.
Grande parte do trabalho de aplicação do QbD em P&D é dedicado ao entendimento do medicamento em desenvolvimento e inclui a coleta e organização de dados. O processo de desenvolvimento de medicamentos desde o conceito até um lote clínico inicial gera uma quantidade significativa de dados e esses dados devem ser gerenciados de forma eficaz ao longo do tempo em várias disciplinas funcionais.
O ambiente rico em dados criado ao aplicar QbD à P&D farmacêutica exige um sistema de gestão do conhecimento com três componentes principais: bancos de dados confiáveis, sistemas de relatórios que apoiam a tomada de decisões e observações científicas sólidas. Vamos examinar cada um deles um pouco mais detalhadamente.
Bancos de dados confiáveis
O desenvolvimento bem-sucedido de medicamentos requer muitos recursos e, sem dúvida, os dados são um dos mais essenciais. A importância dos bancos de dados exige que sejam confiáveis, mas o que constitui um banco de dados confiável? Para mim, bancos de dados confiáveis têm dois recursos principais: integridade e integridade.
A integridade de dados é um tópico de grande interesse em toda a indústria farmacêutica e muitos foram escritos sobre isso. Nos últimos vinte anos, nossa capacidade de usar os dados para obter melhores resultados melhorou, assim como a importância de sua integridade. Para tratar da conformidade com cGMP, o FDA emitiu a orientação final para integridade de dados em 2016. Este não se destina a ser um documento sobre integridade de dados, no entanto, criamos gigabytes de dados ao longo do ciclo de vida de um medicamento. Esses dados devem ser precisos e direi simplesmente que garantir a integridade dos dados é uma função de liderança.
A integridade dos bancos de dados significa que deve haver dados corretos em quantidade suficiente para avançar o desenvolvimento, mas não tanto que os recursos sejam gastos desnecessariamente em experimentos sem direção. A integridade do banco de dados requer um perfil de produto alvo de qualidade (QTTP) concluído o mais cedo possível com atributos críticos de qualidade (CQA) bem definidos. Por exemplo, um problema de uniformidade do produto pode ser descoberto cedo, o que impedirá o aumento de escala se a variabilidade não puder ser reduzida. A falta de dados completos pode atrasar, ou mesmo impedir, uma solução necessária para suportar o aumento de escala.
Relatórios informativos para apoiar a tomada de decisões
Existem dois componentes principais para relatar dados para apoiar a tomada de decisão eficaz para avançar no desenvolvimento de medicamentos - relatórios compreensíveis e a presença de tomadores de decisão.
Os relatórios de dados científicos variam de acordo com a organização e o projeto de desenvolvimento, e há muitas boas soluções de tecnologia disponíveis. Recomendo que as organizações de P&D sejam intencionais nas plataformas que escolhem para apoiar seus esforços. Esforços bem-intencionados para capacitar cada cientista com procedimentos separados de sua escolha ou para criar eficiências com relatórios excessivamente padronizados levaram a disfunções. Os líderes de P&D devem equilibrar a necessidade de relatórios padronizados com o apoio ao processo científico. Para ajudar a atingir esse equilíbrio, é importante entender o que os tomadores de decisão precisam e deixar que isso conduza os relatórios.
Ter o tomador de decisões presente parece óbvio, mas participei de muitas reuniões que apresentaram dados valiosos com a intenção de ajudar a avançar em uma decisão sem a presença de um tomador de decisão. Primeiro, vamos definir a autoridade para tomar decisões. Um tomador de decisão é uma ou mais pessoas que têm autoridade para alocar recursos e estabelecer ou modificar prioridades para dar os próximos passos para avançar um projeto de desenvolvimento de medicamentos. A presença de um tomador de decisões é um imperativo que nivela a organização que toma decisões, cria eficiência e reduz muito os atrasos.
Afirmei que o tomador de decisão pode ser mais de uma pessoa. A tomada de decisão colaborativa é comum e, portanto, torna-se importante apresentar os dados ao órgão de decisão coletiva. Além disso, sou chefe de P&D para uma organização de desenvolvimento e fabricação de contrato (CDMO) que fornece serviços para clientes. Nosso modelo de atendimento é colaborativo e transparente, sendo fundamental incluir a tomada de decisão do cliente em nossos esforços.
Habilitando Observações Científicas
Ser capaz de dar significado às observações dos cientistas de P&D é um desafio e é onde a arte do desenvolvimento de medicamentos desempenha um papel proeminente. Não existem modelos estatísticos que nos ajudem a definir tendências nas observações científicas e os esforços para quantificar as observações científicas geralmente são insuficientes. Por exemplo, você pode realmente padronizar a maneira como um cientista descreve como um comprimido se desintegra em uma cesta de dissolução? E se você pudesse, você gostaria?
No cerne do QbD está a capacidade de estudar atributos críticos de material (CMA) e parâmetros críticos de processo (CPP) para formar atributos críticos de qualidade (CQA). Este nível de discernimento requer cientistas experientes que estão confiantes em fazer julgamentos críticos. Os cientistas que fazem essas ligações devem compreender as especificações pretendidas do produto acabado, o propósito terapêutico final do medicamento e os pacientes aos quais ele se destina.
Uma organização de P&D que aplica QbD de maneira eficaz requer uma abordagem de liderança e cultura organizacional distintas que possam articular os resultados pretendidos e, então, dar à equipe a flexibilidade e o espaço para buscar esses resultados. Nada demonstra mais a força de trabalho moderna baseada no conhecimento do que uma equipe de P&D farmacêutica trabalhando da maneira que descrevi.
O QbD começa no estágio de P&D do desenvolvimento de medicamentos e requer recursos avançados de gerenciamento de conhecimento com três componentes principais: dados confiáveis, relatórios claros que apoiam a tomada de decisões e uma equipe de cientistas experientes que exercem julgamento para alcançar os resultados pretendidos. A P&D baseada em QbD como eu descrevi pode ajudar a agilizar o processo de desenvolvimento de medicamentos para evitar atrasos e fornecer melhores resultados mais rapidamente aos pacientes.
Shawn Watson é responsável por todo o desenvolvimento de produtos, incluindo formas de dosagem estéreis, não estéreis, orais e tópicos, bem como o desenvolvimento de métodos analíticos na Pii. Ele tem mais de vinte anos de experiência em liderança na indústria farmacêutica em organizações de fabricação e desenvolvimento de produtos especiais, genéricos e de contrato (CDMOs). Sua riqueza de conhecimento abrange Pesquisa e Desenvolvimento, Qualidade e Serviços Técnicos.
Antes de ingressar na Pii, Shawn atuou como vice-presidente de qualidade e operações laboratoriais da Lupin Pharmaceuticals, vice-presidente de conformidade da Sigmapharm Laboratories e supervisor de P&D analítico da Teva Pharmaceuticals. Além disso, ele ocupou vários outros cargos importantes, incluindo vice-presidente de qualidade, diretor sênior de serviços de química e manufatura e gerente sênior de pesquisa e desenvolvimento analítico. A paixão de Shawn por trabalhar na indústria farmacêutica começou com seu primeiro emprego como químico de controle de qualidade com um CDMO e incluiu trabalhar com Pii. Shawn obteve um mestrado em administração de empresas pela Fox School of Business da Temple University, um mestrado em química pela Villanova University e um bacharelado em ciências em química e biologia pela Heidelberg University.
Sobre Pii
Pharmaceutics International, Inc. (Pii) é uma organização de desenvolvimento e fabricação de contrato (CDMO) com uma paixão por resolver problemas de forma eficiente com os mais altos padrões de qualidade.
O campus de Pii's Hunt Valley, em Maryland, inclui 70 suítes de fabricação com 4 linhas de enchimento asséptico integradas, proporcionando qualidade, segurança e eficiência. Nossos profissionais têm ampla experiência com compostos de moléculas pequenas e grandes, desenvolvimento e fabricação de medicamentos parenterais complexos, formulações de liberação prolongada, medicamentos injetáveis não aquosos e liofilização. Saiba mais em https: //www.pharmint. com /
quinta-feira, 18 de fevereiro de 2021
Fornecedor de equipamentos de manufatura farmacêutica destaca capacidade de sala de aula ampliada e demonstrações de produtos
A Federal Equipment Company, fornecedora estabelecida de equipamentos de embalagem e fabricação de produtos farmacêuticos, começou 2018 com uma sala de aula 20% maior para o “Processo de Fabricação” e outros treinamentos da Techceuticals. A primeira oferta de 2018 foi “The Manufacturing Process”, que oferece uma experiência prática única para aprender sobre a fabricação de produtos farmacêuticos. Projetado para todos na organização de fabricação farmacêutica - operadores, supervisores, equipe de P&D, formuladores, QA / QC, engenharia, compras, profissionais de manutenção, vendas e até mesmo atividades de administração e suporte, este curso abrangente fornece uma compreensão profunda das operações de dosagem sólida independentemente do nível de habilidade.
O curso é ministrado por Michael D. Tousey, Diretor Técnico e CEO da Techceuticals, um provedor de serviços de treinamento e solução de problemas para empresas de fabricação de medicamentos de dosagem sólida que oferece de tudo, desde programas de treinamento completos; procedimentos; processos; consultoria de formulação; e comparação e seleção de equipamentos. Além do curso Techceuticals, fornecedores de equipamentos em destaque demonstraram equipamentos para os participantes. O evento de janeiro apresentou fornecedores de equipamentos renomados da cadeia de suprimentos farmacêuticos, incluindo Robert Bosch Packaging GmbH e Alexanderwerk, Inc.
Robert Bosch Packaging, Inc. demonstrou a operação de um novo enchimento de cápsula GKF Capsylon 705 fornecido especificamente para este treinamento. O GKF Capsylon 705 apresenta estações modulares com sincronização de tempo e é projetado para uma produção de 42.000 cápsulas por hora; adequado para execuções de lote médio e mudanças frequentes de produto. A máquina possui uma tela de toque para operação e administração dos parâmetros de produção e atende aos padrões das Boas Práticas de Fabricação (GMP), permitindo uma rápida troca e limpeza com apenas algumas ferramentas. Todas as peças de contato do produto são facilmente acessíveis e o operador pode limpar a máquina rapidamente.
Alexanderwerk, Inc. fornece equipamentos de granulação e compactação para as indústrias farmacêutica, química e alimentícia. Eles demonstraram a compactação de rolo com um compactador de rolo WP120V PHARMA mantido em fábrica. Este equipamento, projetado especificamente para uso em operações de fabricação farmacêutica, apresenta um design portátil com alimentador de parafuso horizontal com pré-quebra, rolos cantilever, triturador de flocos e pré-granulador e anexos de granulador fino. A unidade é classificada para +/- 5g lote e capaz de operação contínua de 8-40 kg / hora (veja o equipamento aqui: 49356 ).
A Techceuticals tem uma programação de treinamento robusta para 2018, incluindo palestrantes de entidades patrocinadoras como Bosch, Alexanderwerk, IMA, Glatt, O'Hara, GlobePharma e Korsch. Para obter mais informações sobre os próximos eventos de treinamento, visite Techceuticals.com/training . Verifique também a opção de treinamento eletrônico de Operações de Dose Sólida do Techceuticals .
Sobre a Federal Equipment Company
Federal Equipment Company é o seu nome confiável em equipamentos de processamento usados, fornecendo equipamentos usados industriais e de processo confiáveis a preços competitivos para as indústrias farmacêutica, de suplementos e nutricional. Sediada em Cleveland, Ohio, a Federal Equipment Company fornece equipamentos de fabricação farmacêutica confiáveis em todo o mundo por uma fração do custo e prazo de entrega de novos equipamentos. Além disso, a Federal Equipment Company oferece uma variedade de serviços para avaliar, comprar ou liquidar equipamentos e instalações excedentes.
Considerações técnicas para desenvolver sólidos orais (Parte 2)
Que considerações técnicas devem estar em primeiro plano em seu projeto de sólidos orais? Esta série de duas partes apresenta quatro especialistas em sólidos orais da rede global do Pfizer CentreOne e organizações parceiras que discutem as considerações técnicas a serem consideradas. Na segunda parte da série, nossos especialistas consideram os efeitos do design do equipamento e diferentes estratégias de tablet.
1) Considere o design do seu equipamento [Mike Tousey, CEO da Techceuticals]
Excipiente e informações básicas de API precisam ser obtidas para entender qual equipamento é mais adequado para a fabricação em grande escala. Isso inclui a compreensão da morfologia, do teor de umidade e do nível de controle que afetaria o fluxo e a capacidade de fabricação e, por fim, a vida útil do produto. O nível de dosagem e os problemas de qualidade geralmente decorrem da capacidade dos ingredientes misturados de fluir e controlar a potência da dosagem e isso depende de como os materiais interagem e da exposição que obtêm com o tempo.
O design do equipamento para o seu projeto de sólidos orais deve ser considerado no início do ciclo de desenvolvimento, com atenção aos recursos em escala real. Os formuladores devem garantir que os parâmetros do processo usados durante o desenvolvimento sejam transferíveis para equipamentos em escala comercial. O trabalho em pequena escala deve replicar o treinamento de processo em escala real pretendido o mais próximo possível, de modo que o aprendizado possa informar o projeto de equipamento em escala real e ser prontamente escalonável.
Se você investiu tempo adequado na compreensão da API e dos excipientes e suas interações em pequena escala, colherá os benefícios e economizará tempo mais adiante. Com um trabalho de teste bem projetado e cientificamente justificado, eficiências no gerenciamento de tempo, custo e risco podem ser feitas por meio da redução do trabalho necessário em escala total. Essa compreensão do API e da química do excipiente também cria a base para um processo de limpeza robusto.
2) Avalie sua estratégia para cada operação de unidade no início [Kieran Coffey, líder de serviço técnico da unidade de Newbridge da Pfizer na Irlanda]
Na fabricação de sólidos orais, há muitas operações unitárias diferentes. Não existe uma abordagem de tamanho único para todos e é excessivamente simplista pensar que uma estratégia funcionará independentemente do composto. Uma compreensão da operação de cada unidade (e estratégias alternativas) é necessária como uma linha de base para qualquer pessoa que trabalhe no desenvolvimento ou transferência de formulação. Em um alto nível, porém, o objetivo de qualquer pessoa que desenvolva uma estratégia para cada operação de unidade é ser capaz de avaliar tecnicamente o processo usando os seguintes princípios-chave:
Compreender as entradas do processo e remoção de variação
Compreensão do processo
Modelagem e controle de processos
Entradas do processo - Independentemente da operação ou aplicação da unidade, as entradas do processo devem ser detalhadas e caracterizadas. A variação deve ser removida e, onde isso não for possível, as entradas devem ser controladas. As entradas não se limitam aos materiais da primeira etapa do processo. Cada operação da unidade fornece as entradas para a atividade downstream. Portanto, a caracterização e o controle das saídas em cada etapa é fundamental para o controle das entradas da próxima etapa. Testes em pequena escala também permitirão que o espaço de design seja construído para esses atributos materiais.
Compreensão do processo - o que exatamente está acontecendo durante a operação de cada unidade. Uma dica que dou aos recém-formados quando eles começam a trabalhar no centro de desenvolvimento de Newbridge é imaginar que você é a partícula / tablet - o que você vê? Em uma operação de mistura, a distância percorrida por cada partícula é fundamental, para as operações de revestimento, a zona de pulverização, a mistura e o equilíbrio termodinâmico são de grande importância. Ao identificar os parâmetros que têm o maior impacto nas partículas de uma operação, você define os pontos de controle críticos e o espaço de design do seu processo.
Modelagem e controle de processos - Por meio do controle de entradas e dos parâmetros críticos para a operação de uma unidade, pode-se criar um modelo que aumenta muito a taxa de sucesso das operações de escalonamento e transferência. Construir um modelo que prevê o resultado de um processo permite que você mantenha uma estratégia de controle de processo rígida. Ligeiras flutuações nos parâmetros críticos podem identificar desvios do processo antes que afetem o produto. Isso pode ser difícil, especialmente para operações de unidade complexas, mas o tempo gasto nessa tarefa durante o desenvolvimento inicial em pequena escala e a transferência é compensado muitas vezes durante o ciclo de vida do produto.
Antes que novos medicamentos possam chegar aos pacientes que deles precisam, há uma grande quantidade de P&D que é realizada para garantir a segurança e eficácia dos medicamentos. Da pesquisa preliminar de descoberta de medicamentos ao desenvolvimento e progressão de medicamentos por meio de testes clínicos, antes de finalmente chegar ao mercado, o lançamento de uma nova terapia é o culminar de mais de 10 anos de trabalho árduo.
Apesar dos esforços extraordinários e do investimento financeiro significativo por trás de cada candidato a medicamento, apenas uma pequena fração conseguirá passar por testes clínicos - menos de 10%. A principal razão para isso é a baixa solubilidade e biodisponibilidade de candidatos a drogas. Mais de 40 por cento das novas entidades químicas (NCEs) desenvolvidas na indústria farmacêutica exibem baixa solubilidade aquosa, e esse problema é agravado por uma tendência para moléculas hidrofóbicas mais complexas.
Criando um caminho para novos candidatos a drogas
Há uma grande necessidade de tecnologias inovadoras que possam melhorar a taxa de sucesso de novos candidatos a medicamentos e abrir caminho para que novas terapias cheguem ao mercado. Avanços recentes na tecnologia de engenharia de nanopartículas prometem realizar exatamente isso. Ao aumentar a área de superfície dos compostos de drogas, a solubilidade e a biodisponibilidade podem ser bastante melhoradas. Outro benefício importante oferecido pela engenharia de nanopartículas é a redução da dose necessária para o efeito terapêutico, o que reduz os custos de fabricação e o desperdício.
A tecnologia de engenharia de nanopartículas da Nanoform's Controlled Expansion of Supercritical Fluids (CESS ®) cria nanopartículas uniformes a partir da solução por meio de uma técnica de recristalização 'bottom-up'. Usando este método, é possível criar nanopartículas com tamanho inferior a 200 nm e, ocasionalmente, tão pequeno quanto 10 nm. Este é um grande avanço. Ao diminuir o tamanho das partículas - e, por extensão, aumentar a solubilidade e a biodisponibilidade - nessa medida, a tecnologia não só promete aprimorar novas terapias, mas também abre a possibilidade de que novos medicamentos anteriormente descartados devido a problemas de solubilidade possam ser revisitados.
Estabelecendo as bases para o futuro
Em reconhecimento ao poder da tecnologia da Nanoform, a empresa adquiriu recentemente o status de GMP para fabricar um medicamento experimental para uso em testes em humanos. Para impulsionar ainda mais sua tecnologia, Nanoform também passou por um IPO de grande sucesso na Suécia e na Finlândia, durante o qual as ações da empresa foram subscritas em excesso. O lançamento da Nanoform como uma sociedade anônima resultou em mais US $ 80 milhões em investimentos. Com base nessa base sólida, a Nanoform pode agora esperar apoiar um número crescente de projetos de desenvolvimento de medicamentos e ajudar os pacientes a obter acesso global a terapias que mudam vidas mais rapidamente.
terça-feira, 9 de fevereiro de 2021
Secagem por spray para compressão direta de produtos farmacêuticos
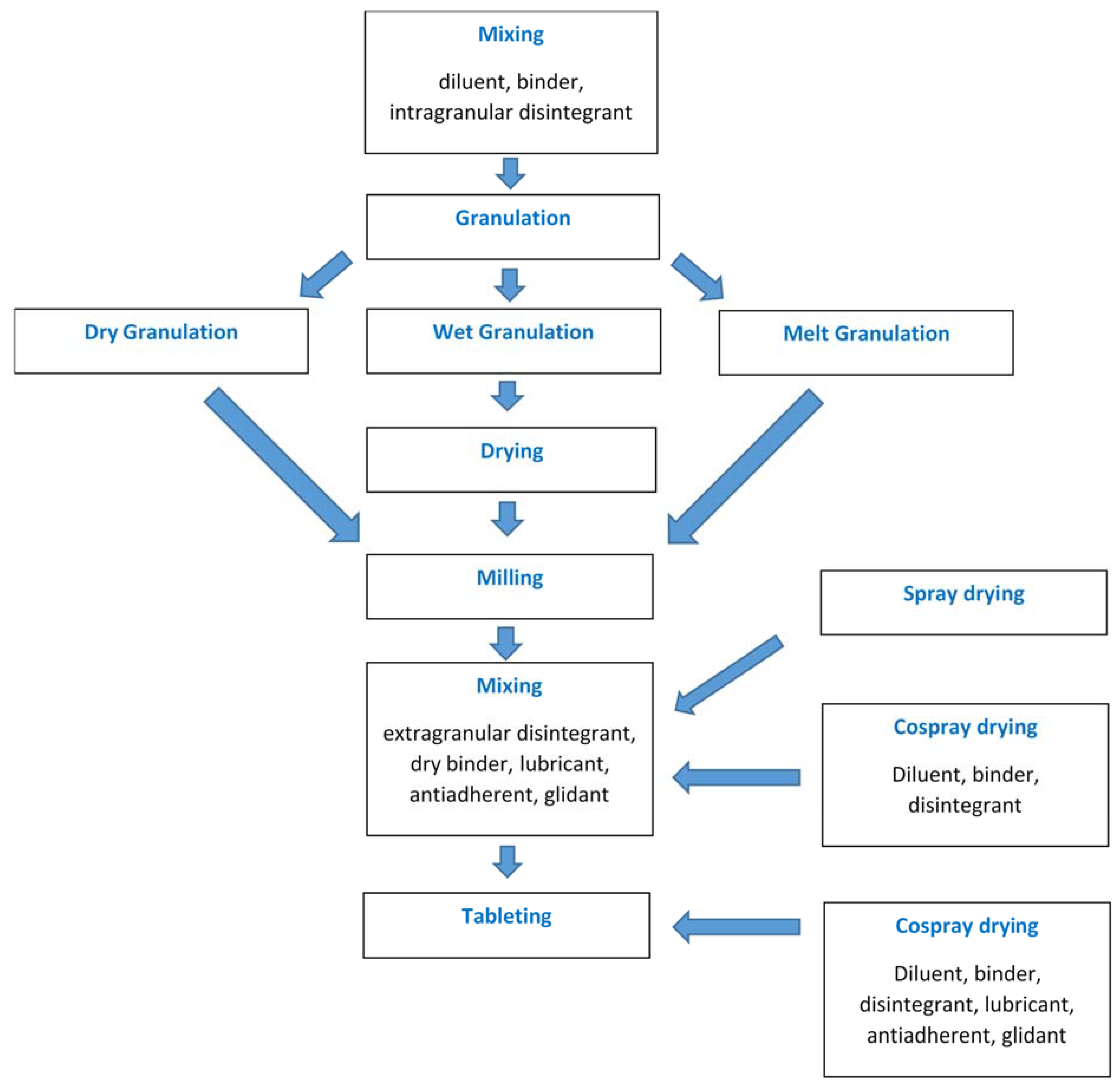
Operações unitárias empregadas na produção de comprimidos via granulação ou spray: secagem conjunta
Comprimir por compressão direta (DC) é uma das abordagens de fabricação de medicamentos mais simples e econômicas. No entanto, a maioria dos ingredientes farmacêuticos ativos (APIs) e excipientes não possuem as propriedades de compressão e fluxo necessárias para atender às necessidades de prensas industriais de comprimidos de alta velocidade.
Portanto, a maioria dos APIs de DC e excipientes são modificados por meio de técnicas de engenharia de partículas de processamento / coprocessamento para aumentar suas propriedades. A secagem por pulverização é uma das técnicas mais comumente empregadas para preparar graus DC de APIs e excipientes com vantagens importantes. Esta revisão tem como objetivo apresentar uma visão geral dos DC APIs e excipientes comercialmente comercializados e preparados em investigação, produzidos por secagem por pulverização.
Visão geral interessante sobre excipientes nesta publicação do Capítulo 5:
5. Excipientes secos por spray de compressão direta
5.1. Lactose seca em spray O primeiro excipiente spray-dried comercialmente disponível para compressão direta foi a lactose, introduzida em 1956 [50]. A secagem por spray resultou em um produto final que compreendia 80-90% de partículas cristalinas de α-lactose mono-hidratada aglomeradas em unidades esféricas com a ajuda do material amorfo restante. O componente amorfo foi produzido a partir da lactose dissolvida na pasta de alimentação. A lactose seca por spray geralmente contém 9-12% de β-lactose, predominantemente na matriz amorfa [51]. Os aglomerados secos por pulverização exibiram fluidez e compressibilidade aprimoradas em comparação com o monohidrato de α-lactose 100% amorfo ou cristalino. O excelente fluxo resultou do grande tamanho de partícula e formato esférico dos aglomerados. Por outro lado, a fração amorfa presente contribuiu para a compressibilidade superior em virtude do maior grau de deformação plástica e melhor ligação [52]. Em teoria, a compactação da lactose spray-dried combina a fragmentação e a deformação plástica do monohidrato cristalino e das frações amorfas, respectivamente [50,53]. No entanto, o mecanismo de consolidação da lactose também depende do tamanho de sua partícula. O diâmetro de transição frágil-dúctil da lactose é de 45 µm, mas quando o tamanho da partícula é menor do que isso, a deformação plástica pode ser o principal mecanismo de consolidação da lactose seca por pulverização [51]. o mecanismo de consolidação da lactose também depende do tamanho de sua partícula. O diâmetro de transição frágil-dúctil da lactose é de 45 µm, mas quando o tamanho da partícula é menor do que isso, a deformação plástica pode ser o principal mecanismo de consolidação da lactose seca por pulverização [51]. o mecanismo de consolidação da lactose também depende do tamanho de sua partícula. O diâmetro de transição frágil-dúctil da lactose é de 45 µm, mas quando o tamanho da partícula é menor do que isso, a deformação plástica pode ser o principal mecanismo de consolidação da lactose seca por pulverização [51]. As características de compressão direta da lactose seca por pulverização inicial foram melhoradas ao longo dos anos, otimizando principalmente dois fatores: a porcentagem de conteúdo amorfo e o tamanho da partícula primária de partículas secas por pulverização de mono-hidrato de α-lactose [54,55]. Por exemplo, a otimização do conteúdo amorfo é reivindicada para explicar a melhor compactabilidade do FlowLac® 90 grau em comparação com o FlowLac® 100 seco por pulverização regular [56]. Diferentes graus de lactose seca por pulverização estão disponíveis comercialmente, diferindo no desempenho de comprimidos e na sensibilidade à umidade ambiental durante o armazenamento. Neste contexto, Pharmatose® DCL 14, um grau de lactose seco por pulverização aprimorado produzido a partir de partículas primárias menores de mono-hidrato de α-lactose, foi comparado com o grau de lactose seco por pulverização regular Pharmatose® DCL 11. Ambos os tipos apresentaram fluxo semelhante, mas compactação diferente comportamento. Pharmatose® DCL 14 foi significativamente mais compactável, o que foi atribuído à maior área de superfície total fornecida pelas partículas primárias menores [51]. A compactabilidade de Pharmatose® DCL 14 foi preservada após armazenamento dos comprimidos a 58% ou 33% UR por 21 dias em comparação com os comprimidos iniciais. Curiosamente, a força dos comprimidos Pharmatose® DCL 11 aumentou após a exposição às condições de alta umidade relativa no mesmo período. A recristalização do conteúdo amorfo nos comprimidos e a formação de pontes foram considerados os motivos para o aumento da força do comprimido. Por outro lado, a pré-exposição do pó à umidade antes da compressão reduziu a compactabilidade de ambos os graus, que foi atribuída à conversão do conteúdo amorfo em cristalino antes da compressão [57]. Finalmente, embora não possa ser generalizado, as qualidades regulares de diferentes fabricantes parecem ter qualidades semelhantes. Por exemplo, os graus regulares de secagem por pulverização de lactose FlowLac® 100 e Pharmatose® DCL 11 foram encontrados para exibir fluidez, compressibilidade e comportamento de compactação semelhantes [58]. A recristalização do conteúdo amorfo nos comprimidos e a formação de pontes foram considerados os motivos para o aumento da força do comprimido. Por outro lado, a pré-exposição do pó à umidade antes da compressão reduziu a compactabilidade de ambos os graus, que foi atribuída à conversão do conteúdo amorfo em cristalino antes da compressão [57]. Finalmente, embora não possa ser generalizado, as qualidades regulares de diferentes fabricantes parecem ter qualidades semelhantes. Por exemplo, os graus regulares de secagem por pulverização de lactose FlowLac® 100 e Pharmatose® DCL 11 foram encontrados para exibir fluidez, compressibilidade e comportamento de compactação semelhantes [58]. A recristalização do conteúdo amorfo nos comprimidos e a formação de pontes foram considerados os motivos para o aumento da força do comprimido. Por outro lado, a pré-exposição do pó à umidade antes da compressão reduziu a compactabilidade de ambos os graus, que foi atribuída à conversão do conteúdo amorfo em cristalino antes da compressão [57]. Finalmente, embora não possa ser generalizado, as qualidades regulares de diferentes fabricantes parecem ter qualidades semelhantes. Por exemplo, os graus regulares de secagem por pulverização de lactose FlowLac® 100 e Pharmatose® DCL 11 foram encontrados para exibir fluidez, compressibilidade e comportamento de compactação semelhantes [58]. que foi atribuída à conversão de conteúdo amorfo em cristalino antes da compressão [57]. Finalmente, embora não possa ser generalizado, as qualidades regulares de diferentes fabricantes parecem ter qualidades semelhantes. Por exemplo, os graus regulares de secagem por pulverização de lactose FlowLac® 100 e Pharmatose® DCL 11 foram encontrados para exibir fluidez, compressibilidade e comportamento de compactação semelhantes [58]. que foi atribuída à conversão de conteúdo amorfo em cristalino antes da compressão [57]. Finalmente, embora não possa ser generalizado, as qualidades regulares de diferentes fabricantes parecem ter qualidades semelhantes. Por exemplo, os graus regulares de secagem por pulverização de lactose FlowLac® 100 e Pharmatose® DCL 11 foram encontrados para exibir fluidez, compressibilidade e comportamento de compactação semelhantes [58].
Representação esquemática do instrumento de secagem por spray. BUCHI Labortechnik AG [315,2 Celulose microcristalina (MCC) A celulose microcristalina (MCC) é outro excipiente seco por pulverização amplamente utilizado. Embora possa ser sintetizado por diferentes processos, a hidrólise de ácido mineral é o método de escolha para produzir α-celulose pura. Primeiro, a celulose é parcialmente despolimerizada e em uma etapa seguinte é neutralizada e seca por pulverização [59,60]. O produto final é um pó branco, insípido e inodoro. Diferentes graus de várias distribuições de tamanho de partícula e conteúdo de umidade estão disponíveis, os quais podem ser produzidos controlando a aglomeração por meio da manipulação das condições de secagem por pulverização. Além disso, polpas de celulose específicas possibilitaram a fabricação de graus de MCC que exibem alta densidade aparente [61]. Alguns dos produtos disponíveis comercialmente são Avicel® ,Vivapur® e Emcocel® . Para obter informações adicionais sobre a preparação de vários graus de MCC e suas propriedades e aplicações, o leitor pode consultar um capítulo de livro recente de Chaerunisaa et al. [62] e uma extensa revisão por Thoorens et al. [61].
5,3. Sais de cálcio secos por pulverização Três diferentes sais de cálcio foram desenvolvidos como materiais granulares por secagem por pulverização para melhorar suas propriedades de compactação e fluxo. O primeiro é o lactato de cálcio pentahidratado (Puracal® DC), um material cristalino quebradiço que exibe baixa sensibilidade à velocidade de compactação. No entanto, possui baixa tendência à fragmentação, o que o torna um enchimento sensível ao lubrificante [63]. A desintegração e a dissolução de formulações de comprimidos de protótipo usando lactato de cálcio penta-hidratado sozinho ou com MCC foram rápidas e completas [63]. O próximo excipiente de sal de cálcio é fosfato dicálcico anidro seco por pulverização ( Fujicalin®) Suas propriedades foram investigadas exaustivamente e comparadas com o fosfato dicálcico anidro regular correspondente; possui menor tamanho de partícula com maior área superficial específica, porosidade e compactabilidade, mas fluidez semelhante [64,65]. Devido à sua alta fragilidade, Fujicalin® exibiu estabilidade contra excesso de lubrificação com estearato de magnésio, o que foi demonstrado para diferentes misturadores e tempos de mistura [66]. O terceiro excipiente à base de sal de cálcio é o fosfato tricálcico seco por pulverização ( Tri-Cafos® 500. Tem uma estrutura esponjosa e, portanto, uma grande área de superfície específica. Ao contrário dos outros sais de cálcio diretamente compressíveis, consolida principalmente por deformação plástica Quando usado como um co-diluente, resulta em aumento da porosidade e desintegração mais rápida do comprimido [67].
5,4 Açúcares secos em spray e polióis Os açúcares secos em spray e polióis estão ganhando cada vez mais atenção como enchimentos em comprimidos mastigáveis, chupáveis, efervescentes e de desintegração oral. Os exemplos incluem sacarose (Compressuc® PS), maltose (Advantose® 100), manitol ( Parteck® M , Pearlitol® SDe Mannogem®) e sorbitol (Neosorb® XTAB, Parteck® SI). Entre estes, o manitol possui uma gama de propriedades interessantes que o tornaram um dos enchimentos mais usados para as formas de comprimidos acima mencionadas [68]. Além da doçura, possui um calor de solução negativo liberando uma sensação de resfriamento na boca. Além disso, não é higroscópico, embora seja muito solúvel em água, oferecendo excelente estabilidade e compatibilidade com medicamentos [69]. O manitol seco por pulverização está disponível comercialmente em dois graus: o 100 ou EZ e o 200 ou XL com o primeiro tendo um tamanho médio de partícula menor [70]. Ao contrário da lactose, a secagem por spray não afeta sua cristalinidade, sendo o produto final composto por cristais β (Parteck®) ou uma mistura de cristais de manitol α e β (Pearlitol® e Mannogem®) [71,72]. Recentemente, as propriedades do pó e do comprimido de várias marcas e graus de manitol seco por pulverização foram comparadas com os graus granulados disponíveis comercialmente [70,72]. Exceto por um pequeno aumento no tempo de desintegração observado após armazenamento de uma semana (25 ° C / 75% UR), os comprimidos de graus de manitol seco por pulverização mostraram estabilidade física superior sobre os graus à base de granulação. O Neosorb® XTAB é uma forma em pó de sorbitol desenvolvida pela Roquette como um enchimento amigo dos dentes para fazer comprimidos sem açúcar [73]. Sorbitol instantâneo (Parteck® SI, Merck) é um grau cristalino de sorbitol de baixa higroscopicidade de fluxo livre e seco por pulverização com compressibilidade melhorada e alta capacidade de adsorção para a preparação de misturas ordenadas devido aos seus cristais filamentosos entrelaçados frouxamente empacotados, orientados aleatoriamente [ 74]. Advantose® 100 é uma forma de pó não higroscópico de fluxo livre diretamente compressível do dissacarídeo maltose. Apesar de sua natureza cristalina e fragmentação durante a consolidação, sua compactabilidade mostrou ser afetada por lubrificantes em uma faixa de forças de compressão [75]. Compressuc® PS é um grau de sacarose spray-dried que está em conformidade com a monografia “sacarose” da USP / NF e Ph. Eur. [76].
5.5. Amido de arroz seco por pulverização Era-tab® e Primotab ET® são excipientes com base em partículas de amido de arroz aglomerado que eram usados como graus secos por pulverização disponíveis comercialmente. A utilidade do amido de arroz seco por spray para compressão direta foi avaliada em relação a vários enchimentos em termos de propriedades físicas e capacidade de compressão. Era-tab® demonstrou possuir excelente fluidez, compactabilidade, bem como propriedades de desintegração e dissolução. Dentre suas desvantagens, as mais importantes são o potencial de diluição moderado e a sensibilidade do lubrificante [77,78]. No entanto, este último é menor quando comparado a outros produtos de amido, e é considerado suficiente para formulações de comprimidos [79].
Representação esquemática da cinética de secagem de gotas de uma única gota durante o processo de spraydrying. Reproduzido de Boel et al. [33]5,6. Produtos em investigação Além de estudos sobre excipientes secos por pulverização comercializados, foi relatada a investigação do efeito do processo de secagem por pulverização nas propriedades mecânicas e retrabalho de alguns polímeros conhecidos. Os principais estudos de pesquisa sobre excipientes secos por pulverização experimental ou co-pulverizados estão resumidos na Tabela 2. Succinato de acetato de hipromelose seco por pulverização (HPMCAS) de soluções orgânicas exibiu maior dureza e menor limite de rendimento fora do molde [80], mas maior resistência à tração de compactos do que material não processado, indicando capacidade aprimorada de formar compactos fortes no carregamento [49,80]. Rege et al. [81,82] quitinosanos secos por pulverização que foram submetidos a N-desacetilação e despolimerização. Em comparação com o produto seco em bandeja, as partículas de quitinosana secas por pulverização foram mais isodiamétricas e exibiram melhor fluidez. Além disso, as propriedades de compactação de sua mistura com a tetraciclina, usada como medicamento modelo, foram superiores. Kolakovic et al. investigou as propriedades físicas e mecânicas de nanofibras de celulose secas por pulverização em comparação com dois graus comerciais de MCC, Avicel® PH101 e Avicel® PH102. Os resultados mostraram que a celulose nanofibrilada seca por pulverização tem uma melhor fluidez do que Avicel® PH101 e sua adição ao Avicel® PH102 melhorou a fluidez das misturas resultantes. Comprimidos com uma droga modelo (paracetamol) foram preparados com sucesso em uma prensa excêntrica. No entanto, em comparação com a celulose microcristalina, a celulose nanofibrilada seca por pulverização era mais frágil, mas menos deformável e compactável [83]. Os resultados dos estudos acima mostram que a secagem por pulverização diminuiu a compressibilidade, mas aumentou a fragilidade dos excipientes à base de carboidratos. Uma vez que o resultado da compactação também depende do estágio inicial do rearranjo das partículas antes da formação do compacto, e uma vez que as partículas secas por pulverização alcançam um empacotamento mais uniforme e um rearranjo mais fácil das partículas, o efeito geral da secagem por pulverização na capacidade de compressão deve ser balanceado considerando os efeitos individuais fluidez e comportamento de compressão.
6. Excipientes Secos Co-Spray de Compressão Direta
6.1. Com base na lactose
6.1.1. Lactose-Celulose (Cellactose®) Cellactose® 80é composto por 75% de α-Lactose mono-hidratada e 25% de celulose em pó (proporção 3: 1) mistura seca por pulverização conjunta. Seu ângulo de repouso relatado é de 34 °, a densidade aparente de 0,37 g / cm3 e a densidade compactada de 0,49 g / cm3 [84]. Segundo o fabricante [84], é projetado como excipiente para a fabricação de comprimidos por compressão direta devido a sua fluidez superior, reduzida tendência de segregação e compressibilidade. Casalderrey et al. [85] comparou as propriedades de uma proporção de 3: 1 de combinação de α-Lactose mono-hidratada / celulose microcristalina processada por granulação a seco ou extrusão / esferonização com aquelas de pó de Cellactose® de tamanho de partícula semelhante. Cellactose® mostrou melhor esfericidade e fluidez do que a mistura de granulação seca (índice de Carr de 24% em comparação com 37%). Além disso, Cellactose® melhorou as propriedades mecânicas, mas uma desintegração muito mais pobre do que os comprimidos das outras misturas comprimidos à mesma alta pressão de punção. A força e a resistência à água de comprimidos de Cellactose® bem compactados foram atribuídas à distribuição espacial de celulose e lactose em partículas de Cellactose®, ao invés do conteúdo de β-lactose ou características estruturais extra-particulares [85]. Arida e Al-Tabakha [86] relataram compactabilidade superior de Cellactose® sobre a mistura física de contrapartida.
6.1.2. Lactose – MCC (MicroceLac® 100) MicroceLac® 100é uma mistura seca por pulverização conjunta de 75% de α-Lactose mono-hidratada e 25% de celulose microcristalina [87]. De acordo com o folheto do produto (graus de lactose coprocessada da MEGGLE para compressão direta: MicroceLac® 100), possui um índice de Carr de 21%, ângulo de repouso igual a 34 ° e densidade aparente de 0,46 g / cm3 e é reivindicado ter boas propriedades de fluidez e compactação, tornando-o adequado para compressão direta. Em comparação com sua mistura física de contrapartida, MicroceLac® 100 apresentou melhor fluidez e resistência mecânica do comprimido [88,89], desintegração e dissolução mais rápida do medicamento (f2 = 16,61) [89]. No entanto, era mais suscetível à cristalização induzida por água devido à instabilidade termodinâmica e à cristalização do conteúdo amorfo em uma forma cristalina estável [88].
6.1.3. Lactose-Amido (StarLac®) StarLac®é um material seco por pulverização conjunta composto por 85% de α-lactose mono-hidratada e 15% de amido de milho branco fabricado pela Meggle Pharma. Segundo o fabricante, seu índice de Carr é de 19,4% e o ângulo de repouso é de 29 ° [91]. O índice de Carr mais baixo em comparação com Cellactose® e MicroceLac® 100 pode ser atribuído à maior esfericidade e superfície mais lisa das partículas (Figura 5). A pressão de escoamento Heckel relatada é menor para StarLac® do que para FlowLac®, indicando maior plasticidade [90,92]. Hauschild e Picker investigaram as propriedades de compressão de StarLac® e compararam-no com FlowLac® (lactose spray-dried) e uma mistura física correspondente de FlowLac® e amido de milho. Eles concluíram que StarLac® tem boa compactabilidade e propriedades de compressão semelhantes ao FlowLac®. Além disso,
6.1.4. Lactose-MCC-Starch (CombiLac®) CombiLac®é composto por 70% de α-lactose mono-hidratada, 20% de celulose microcristalina e 10% de amido de milho branco nativo [94]. Mužíková et al. [95] estudaram as propriedades dessa mistura ternária seca por pulverização. Eles compararam a fluidez, compactabilidade e propriedades do comprimido do CombiLac® com a mistura física correspondente. Eles descobriram que o CombiLac® tinha uma razão de Hausner mais baixa, indicando melhor empacotamento e fluidez, e melhor capacidade de compressão do que a mistura física. Além disso, a compactação do CombiLac® foi mais sensível à presença de lubrificantes na formulação, o que é explicado pela menor propensão à fragmentação do excipiente principal frágil (lactose) quando co-spray dry com os co-excipientes plásticos (amido e MCC ) em comparação com a mistura física correspondente. Bowles et al. [96] caracterizou o comportamento de compressão para uma variedade de excipientes coprocessados. Os resultados revelaram que o CombiLac® tinha índice de Carr mais baixo, melhor ejetabilidade, desintegração compacta ligeiramente mais rápida (<60 s), mas perfis de compressibilidade, compactabilidade e capacidade de compressão semelhantes em comparação com o MicroceLac® 100.
6,2 Com base em MCC
6.2.1. MCC – Manitol (Avicel® HFE) Celulose microcristalina-Manitol ( Avicel® HFE) é um material seco por pulverização conjunta composto de 90% de celulose microcristalina e 10% de manitol [98]. Avicel® HFE-102 é relatado como tendo um tamanho de partícula nominal de 100 µm, ângulo de repouso de 30,67 °, densidade aparente em torno de 0,41 g / cm3 e índice de Carr de 19,19% [98,99]. Ele mostrou comportamento de compressão semelhante e desintegração ligeiramente mais rápida de compactos em comparação com Avicel® PH-102[90,96]. Além disso, os comprimidos comprimidos a uma pressão de 78 MPa eram fortes com resistência à tração entre 5 e 6 MPa, friabilidade de 0,01% e tempo de desintegração 4,03 min [99]. Esta combinação de manitol / celulose microcristalina melhora a palatabilidade e diminui o tempo de desintegração devido à boa solubilidade em água, propriedades de umedecimento e calor negativo da solução de manitol [100]. Portanto, este material coprocessado é adequado para compressão direta de comprimidos mastigáveis.
6.2.2. MCC – Goma de Guar (Avicel® CE-15) Goma de celulose-guar microcristalina (Avicel® CE-15) é um material seco por pulverização conjunta composto de 85% de celulose microcristalina e 15% de goma de guar [98,99]. Possui índice de Carr 19,46%, indicando fluidez intermediária [99]. Avicel® CE-15 é relatado como tendo um D90 de 221,06 µm, uma área de superfície específica de 0,5 m2 / ge um teor de umidade de 4,66% [99]. Também foi relatado que os comprimidos comprimidos a 130 MPa tinham friabilidade de 0,58%, tempo de desintegração 7,27 min e resistência à tração em torno de 2 MPa. Portanto, é adequado para operações de compressão direta, especialmente para comprimidos mastigáveis devido à goma de guar, que fornece uma experiência sensorial adequada ao reduzir a textura do MCC.
6.2.3. MCC – Fosfato dicálcico (Avicel® DG) Avicel® DG é um material seco por pulverização conjunta composto de 75% de celulose microcristalina e 25% de fosfato de cálcio dibásico anidro. É proposto pelo fabricante para a formação de comprimidos de materiais difíceis de comprimir e para granulação a seco [98]. A combinação de plástico e material quebradiço diminui a ligação durante a compactação com rolo, facilitando a quebra em grânulos. Subseqüentemente, durante a formação de comprimidos, tanto a grande quantidade de pequenas partículas do excipiente inorgânico quanto as superfícies das partículas de celulose microcristalina de deformação plástica fornecem área de superfície suficiente para a ligação e a formação compacta [99]. Avicel® DG foi investigado para a formação de comprimidos de DC, e seus comprimidos mostraram desintegração rápida, alta resistência à tração e baixa friabilidade. No entanto, tem fluidez abaixo do ideal com índice de Carr relativamente alto (24,98%) [99].
6.2.4. Celulose microcristalina silicificada Graus de celulose microcristalina silicificada (MCC) Prosolv® SMCC e Avicel® SMCC são materiais secos por pulverização conjunta compostos de 98% de celulose microcristalina e 2% de dióxido de silício coloidal (CSD) [1,98,101]. Cinco graus de Prosolv® SMCC (50, 50 LD, 90, HD 90 e 90 LM) e três graus de Avicel® SMCC (50, 90 e HD 90) estão disponíveis comercialmente que diferem em tamanho de partícula e densidade aparente. MCC silicificado mostrou melhor fluidez [102] e pegajosidade reduzida [103] em comparação com graus não silicificados. Além disso, foi relatado que os compactos de Prosolv® SMCC 90 e Prosolv® SMCC 90 HD tiveram tempo de desintegração abaixo de 1 min a forças de compressão de 3, 3,5 e 4 KN [104]. O comportamento de compactação do Prosolv® SMCC 90 foi estudado e comparado ao MCC [105]. Ambos os excipientes apresentaram comportamento de densificação semelhante para pressões de compressão de até 400 MPa. Além disso, verificou-se que o relaxamento pós-compactação dos comprimidos correspondentes também era semelhante. No entanto, a presença de lubrificante (estearato de magnésio) afetou o relaxamento dos comprimidos SMCC 90, o que foi atribuído a um pequeno efeito negativo do CSD na força de ligação interpartículas do MCC não lubrificado. A resistência mecânica dos comprimidos dos dois excipientes foi semelhante em pressões de compactação de até 400 MPa. A mistura com o lubrificante diminuiu a força para ambos os tipos de comprimidos e o efeito foi mais pronunciado no caso do MCC [105]. Kachrimanis et al. [106] confirmaram que há um ligeiro aumento na força do comprimido, mas um aumento acentuado no tempo de desintegração do Prosolv® em comparação com os comprimidos Avicel® para frações sólidas entre 0,7 e 0,9, que é semelhante à faixa de comprimidos farmacêuticos. Eles também sugeriram que o SiO2 atua como uma barreira ou dreno de umidade para UR de até 52%, mas com UR de 72% os espaços interpartículas tornam-se saturados com umidade, não permitindo a penetração de água no MCC, que não pode atuar como desintegrante, resultando em tempos de desintegração mais elevados . Em 2010, o JRS PHARMA introduziu o Prosolv® EASYtab como um excipiente que cumpre vários recursos desejados para fins de compressão direta [107]. Pode ser simplesmente misturado com grânulos ou graus DC de ingredientes ativos para formar uma mistura RTC [108], mas desintegrante extra deve ser adicionado para produtos contendo alto teor de dispersões sólidas a fim de encurtar o tempo de desintegração [109]. Aljaberi et al. [110] comparou o desempenho de compactação e dissolução de Prosolv® EASYtab com misturas físicas correspondentes de MCC ou SMCC com excipientes complementares (dióxido de silício coloidal, glicolato de amido sódico e estearil fumarato de sódio). Prosolv® EASYtab exibiu compactabilidade, dissolução e estabilidade comparáveis com as misturas físicas MCC ou Prosolv® SMCC correspondentes.
6.3. À base de amido
StarCap1500® StarCap1500® é um material seco por pulverização conjunta composto de 90% de amido de milho e 10% de amido pré-gelatinizado [1]. Mužíková e Eimerová [111] investigaram as energias de compactação, resistência mecânica e tempo de desintegração de compactos StarCap1500®. Eles descobriram que tinham maior resistência à tração do que Starch1500® do mesmo tamanho comprimido nas mesmas condições. No entanto, a resistência compacta do StarCap 1500® foi significativamente reduzida quando misturado com 0,4% de estearato de magnésio, o que deve ser atribuído à tendência de baixa fragmentação, uma vez que o amido é elastoplástico. Além disso, os pesquisadores descobriram que os comprimidos StarCap 1500® tinham um tempo de desintegração significativamente menor em comparação com o Starch®1500, que foi atribuído a uma rede capilar mais eficiente.
6,4 À base de açúcar
6.4.1. Glicose-Dextratos (Emdex®) Glicose monohidratada - Dextrates, NF (Emdex®) é fabricado por JRS PHARMA. De acordo com o fabricante [112], este material seco por pulverização conjunta compreende 95% de monohidrato de glicose e 5% de oligossacarídeos derivados de amido (Dextratos, NF). Tem um tamanho médio de partícula de 200 µm e oferece vários benefícios como agente de enchimento / ligante solúvel em água em formulações de compressão direta. Possui excelentes propriedades de empacotamento e fluxo com razão de Hausner de 1,07 e ângulo de repouso de 30 °. Além disso, a compressão do Emdex® com forças entre 3 kN e 15 kN produziu comprimidos com resistência à tração entre 0,3 MPa e 2,5 MPa. Essas características, além de um sabor adocicado agradável, o tornam adequado para compressão direta de formulações de comprimidos mastigáveis, efervescentes e ingeríveis processados em forças de compressão superiores a cerca de 10 kN [112].
6.4.2. Frutose-Amido (Advantose® FS95) Frutose-amido seco por co-pulverização (Advantose® FS95) é fabricado pela SPI Pharma. Apesar da baixa compressibilidade da frutose, quando ela é co-pulverizada com uma pequena quantidade de amido (5%), o produto da combinação torna-se diretamente compressível [113]. Além disso, Advantose® FS95 possui menor higroscopicidade e distribuição de tamanho de partícula que garante boas propriedades de fluxo com um ângulo de repouso de 12 °. Além do excelente perfil de compressibilidade, apresenta friabilidade reduzida. Devido à sua doçura, é recomendado para uso em comprimidos multivitamínicos mastigáveis para crianças [113].
6.4.3. Sacarose – Maltodextrina – Açúcar Invertido Compressuc® MS é um produto seco por pulverização conjunta composto de sacarose, maltodextrina (2,3% ± 0,5%) e açúcar invertido (1,7% máx.) Com uma estrutura de partícula porosa. Atende ao USP-NF na monografia “Açúcar Compressível”. Tem um diâmetro médio entre 150 e 300 µm, densidade aparente entre 0,53 e 0,61 g / cm3 e densidade compactada entre 0,61 e 0,71 g / cm3. Como é o caso com o grau de sacarose spray-dried (Compressuc® PS), Compressuc® MS é particularmente adequado para a produção de comprimidos mastigáveis e efervescentes [76].
6,5. O CS90 e o MS90 de base inorgânica são aglutinantes de enchimento secos por pulverização comercializados pela SPI Pharma. O CS90 é composto por 90% de carbonato de cálcio e 10% de amido com um tamanho médio de partícula típico de 150–175 µm e densidade compactada de 0,85 g / cm3. MS90 compreende 95% de hidróxido de magnésio e 5% de amido com distribuição de tamanho de partícula e densidade semelhantes ao CS 90. Ambos os excipientes são recomendados para a fabricação de comprimidos antiácidos para mastigar e suplementos minerais. Devido à semelhança em suas propriedades físicas, eles podem ser facilmente misturados e combinados para preparar comprimidos antiácido [114,115].
6,6. Produtos de investigação Wang et al. [116] investigaram as propriedades de compressão de α-lactose mono-hidratada, HPMC e crospovidona. Três graus de tamanho de partícula de lactose (90M, 200M e 450M) foram comparados. Eles descobriram que a secagem por pulverização conjunta usando graus 90M e 200M produziu formulações ideais para a formação de comprimidos. No geral, as resistências à tração dos excipientes secos por pulverização conjunta eram maiores do que as das misturas físicas correspondentes de ambas as composições. A melhoria da compactação foi atribuída à presença de HPMC e à formação de cerca de 30% de lactose amorfa durante a secagem por pulverização conjunta.
Produtos secos por pulverização conjunta de celulose microcristalina com amido de arroz para fins de compressão direta foram relatados por Limwong et al. [117]. Os pesquisadores prepararam as misturas secas por pulverização conjunta com composições de amido de arroz de 90%, 80%, 70%, 60% e 50%. A composição com 70% de amido de arroz foi considerada a mais adequada para compressão direta. O pó seco de pulverização conjunta desta composição tinha um ângulo de repouso de 37,2 ° e índice de Carr de 19,2%. A fluidez era melhor do que vários outros diluentes diretamente compressíveis disponíveis no mercado. Além disso, foram avaliadas as propriedades de comprimidos de 500 mg (Ø 12,7 mm) da mistura seca por pulverização conjunta com 70% de amido de arroz comprimido com carga de 8,8 kN. Os comprimidos produzidos tinham uma resistência média de 188,7 N (DP = 8,6), friabilidade 0,6% e tempo de desintegração 2,56 min (DP = 0,22).
As misturas de celulose microcristalina-carbonato de cálcio secadas em co-pulverização foram patenteadas por Mehra et al. em 1987 [118]. Diferentes proporções dos dois componentes foram sugeridas como combinações possíveis. Foi proposta uma proporção de celulose microcristalina para carbonato de cálcio de 60 a 40. A densidade aparente deste produto estava na faixa de 0,35–0,45 g / cm3, e o pH da pasta aquosa na faixa de 9,5–10. Além disso, o perfil de fluidez e compressibilidade do produto era melhor do que o do MCC e exibia baixa sensibilidade a lubrificantes. Li et al. investigou as propriedades do comprimido de hidroxipropilmetilcelulose (HPMC) com cada um de manitol, amido e hidrogenofosfato de cálcio di-hidratado (DCPD) [119]. Eles empregaram um projeto de centro composto para otimização, empregando HPMC (3,5–10,5%) e conteúdo sólido na alimentação (19–44%) como variáveis independentes. Eles encontraram excipiente coprocessado de DC ideal em composições de HPMC de 7,3%, 7,7% e 7,0%, e conteúdos sólidos na alimentação líquida de 44%, 40% e 32% para manitol, amido e DCPD, respectivamente. Outro trabalho de Fan et al. [120] tentaram melhorar a compactabilidade dos sais biliares colato de sódio (SC) e ácido desoxicólico (DOA) que são usados em formulações como intensificadores de absorção. Eles co-pulverizam SC ou DOA com HPMC adicionado em níveis de 0%, 2,5%, 5% e 10% p / p. Os pós obtidos foram comprimidos em comprimidos (Ø 6 mm) com forças entre 50 e 450 kg, e os produtos secos por pulverização conjunta foram comparados com as matérias-primas e misturas físicas a 10% (p / p) HPMC e 90% (p / w) composição SC ou DOA. Os comprimidos produzidos tinham resistência à tração entre cerca de 0,5 MPa e 6 MPa. Os pós secos de pulverização conjunta de SC e DOC com 10% de HPMC demonstraram os melhores perfis de compactabilidade. A capacidade de compressão melhorada dos pós secos por pulverização foi atribuída ao tamanho de partícula fina e ao estado amorfo dos sais biliares após a secagem por pulverização. Em conclusão, Vanhoorne et al. [46] co-pulverização de manitol seco com PVP em percentagens de 0%, 10% e 20%. A secagem em co-spray de manitol em formulações com 0% e 10% de PVP produziu β e α manitol, respectivamente. No entanto, o manitol de secagem por pulverização em formulações com 20% de PVP produziu principalmente δ manitol. As propriedades de compactação dos pós secos por pulverização conjunta foram comparadas às misturas físicas. Manitol seco em co-pulverização com 20% de PVP deu a maior resistência à tração em todas as pressões aplicadas. Saleh et al. [121] bicarbonato de sódio seco por spray usando PVP e óleo de silicone como aditivos auxiliares. Esses aditivos melhoraram significativamente a compressibilidade do bicarbonato de sódio. As combinações de co-atomização preparadas mostraram excelente compressibilidade e o bicarbonato de sódio não foi transformado em carbonato de sódio. Portanto, as características de compressão superiores do bicarbonato de sódio seco por pulverização conjunta tornam-no um futuro candidato na fabricação de comprimidos efervescentes por compressão direta como fonte de dióxido de carbono Embora a secagem conjunta por pulverização seja normalmente abordada como uma estratégia para melhorar as propriedades dos comprimidos, alguns estudos não relataram nenhuma melhoria ou resultados contrários. A maioria deles relata um impacto negativo na compactabilidade dos excipientes quando os surfactantes estão envolvidos no processo de co-pulverização. Por exemplo, Roberts et al. [122] prepararam aglomerados esféricos secos por pulverização de succinato de acetato de hipromelose (HPMCAS) sozinho ou na presença de 1% ou 3% de lauril sulfato de sódio (SLS). Eles relataram um impacto prejudicial da secagem conjunta com SLS na capacidade de compressão do HPMCAS em comparação com o material não processado. Bergren et al. [123] investigaram o comportamento de compressão de mono-hidrato de α-lactose seca por pulverização conjunta com polivinilpirrolidona (PVP) e polissorbato 80. A concentração de PVP na mistura ternária foi de 25% p / pe a do polissorbato 80 0,1% p / p. As pressões de rendimento de Heckel estimadas variaram entre 94 MPa e 127 MPa para todos os pós secos por pulverização em comparação com 87 MPa para pó de lactose seca por pulverização (lactose amorfa). A presença de polissorbato 80 nos pós secos por pulverização conjunta diminuiu a resistência à tração ao longo da faixa de pressão de compressão aplicada (1–6 MPa). A compactabilidade dos comprimidos de lactose seca por pulverização com PVP foi semelhante à da lactose seca por pulverização sozinha. Observação semelhante sobre o efeito do polissorbato 80, como co-excipiente, na compactabilidade de partículas de lactose amorfa preparadas por secagem por pulverização foi encontrada por Fichtner et al. [124]. Eles relataram uma correlação positiva entre a energia superficial dispersiva de pós secos por pulverização e a resistência à tração de seus compactos preparados em porosidades constantes. A secagem conjunta de lignina com lauril sulfato de sódio (SLS) para melhorar o comportamento de compactação da lignina foi investigada por Solomon et al. [125]. Os pesquisadores prepararam pós secos de pulverização conjunta de lignina que incluíam SLS em níveis de 0%, 5%, 10% e 15%. Embora a pressão de rendimento de Heckel e a recuperação elástica tenham diminuído com o aumento da porcentagem de SLS, foi observada uma diminuição da resistência à tração com o aumento do teor de SLS nas composições, atribuída à adsorção de surfactante na superfície da gota, reduzindo assim a ligação interpartículas e resultando em compactação pobre e nivelamento.
7. Excipientes Secos em Co-spray Multifuncional
7.1. Compressão direta e liberação sustentada
7.1.1. Kollidon® SR Kollidon® SR é um pó seco por pulverização conjunta composto por 80% de acetato de polivinila (MW ~ 450.000), 19% de PVP (K 30), 0,8% de SLS e 0,6% p / p de sílica. É composto por partículas arredondadas com diâmetro médio entre 80 e 100 µm e boa fluidez (ângulo de repouso <30 °) [126]. A temperatura de transição vítrea é baixa (em torno de 40–45 ° C) e, portanto, deforma-se principalmente plasticamente [127,128]. Vários estudos demonstraram sua adequação como formadora de matriz para sustentar a liberação do fármaco por compressão direta [128,129,130,131,132], e a liberação é independente da força de compressão [131,133]. Kollidon® SR e suas misturas com monohidrato de teofilina exibiram maior compactabilidade do que MCC e misturas correspondentes [134].
7.1.2. Produtos em investigação O excipiente co-processado com alginato de sódio e lactose foi produzido por secagem por pulverização de uma solução aquosa de α-lactose monohidratada e alginato de sódio (SA) com conteúdo de SA de até 30%. As partículas de compósito secas por pulverização tinham excelentes propriedades micrométricas como enchimento para compactação direta e boa compactabilidade. Os comprimidos-matriz preparados a partir das partículas compostas mostraram liberação mais lenta do fármaco do que a mistura física de lactose e partículas SA na mesma razão lactose / SA. A liberação foi mais retardada em ácido (pH 1,2) do que em solução neutra (pH 6,8) devido ao inchaço dependente do pH de SA [8,135].
7.2. Compressão direta e desintegração oral
7.2.1. F-MELT ® F-MELT é um produto seco por pulverização conjunta com base nos carboidratos xilitol e manitol, desintegrante (crospovidona e MCC) e componente inorgânico. Está disponível em dois graus, denominados C e M. O primeiro grau contém aluminometasilicato de magnésio ( Neusilin® ) como o componente inorgânico, e o último contém hidrogenofosfato de cálcio anidro ( Fujicalin®) Tem uma boa sensação na boca e é projetado para comprimidos de desintegração oral (ODTs) e comprimidos macios para mastigar. Ele fornece alta estabilidade do medicamento devido à não higroscopicidade e pH neutro, alta fluidez devido às densas partículas isodiamétricas, excelente dureza do comprimido, baixa friabilidade e alta carga de medicamento. Não causa cobertura ou aderência durante a compressão [136,137].
7.2.2. PEARLITOL® Flash PEARLITOL® Flash é uma mistura seca de pulverização conjunta de manitol (80–85%) e amido de milho (15–20%). É composto por partículas isodiamétricas com diâmetro médio de 200 µm e teor de umidade de cerca de 1,8%. PEARLITOL® Flash desintegra-se rapidamente, proporcionando uma textura suave, tornando-o adequado para formulações ODT Farmacêuticas e / ou Nutracêuticas [138].






![Figura 3. Representação esquemática da cinética de secagem de gotas de uma única gota durante o processo de spraydrying. Reproduzido de Boel et al. [33]](https://www.pharmaexcipients.com/wp-content/uploads/2021/02/Figure-3.-Schematic-representation-of-droplet-drying-kinetics-of-a-single-droplet-during-the-spraydrying-process.-Reproduced-from-Boel-et-al.-33.png "Figura 3. Representação esquemática da cinética de secagem de gotas de uma única gota durante o processo de spraydrying. Reproduzido de Boel et al.")