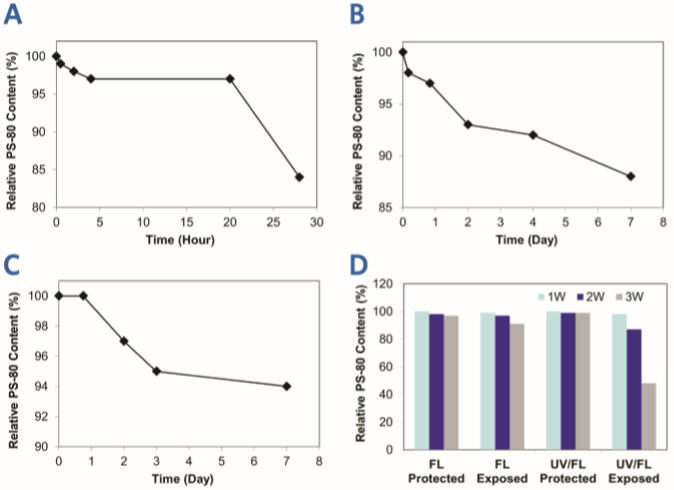
Por Jeff Tremain e Ted Tharp

A liofilização é um processo complexo e importante comumente utilizado para a preservação de terapias biofarmacêuticas. A liofilização pode frequentemente consumir muitos recursos, exigindo muitas horas de trabalho, tempo e investimento, especialmente ao trabalhar com certas classes de drogas biológicas.
Até recentemente, as tentativas de “otimizar” o processo de liofilização eram baseadas em tentativa e erro. Essa abordagem cara foi racionalizada pela maioria dos parceiros biofarmacêuticos e CDMO / CMO como um custo de fazer negócios. Na verdade, em muitos ambientes de fabricação biofarmacêutica, a tentativa e erro em escala piloto com base no know-how ainda é comum e aceita como ponto de partida para muitos processos de liofilização. Mas, mesmo em ambientes onde a substância medicamentosa em si é barata, a abordagem do teste pode representar custos significativos durante a fase de desenvolvimento de um produto. O comprometimento de pessoal, instalações de produção e serviços analíticos no desenvolvimento de produtos em estágio inicial com ciclos de secagem potencialmente longos, pode levar a um lote perdido e produtividade. Isso inevitavelmente interrompe a progressão do pipeline e impede uma fabricação mais lucrativa e eficiente.
O aumento de escala e a transferência de tecnologia dos processos de liofilização continuam sendo um desafio. Mas os avanços recentes baseados em dados na modelagem de computador em estado estacionário e subsequentes práticas de liofilização em escala de bancada estão tendo um impacto positivo na otimização dos processos de liofilização. A dinâmica de fluidos computacional e a análise de atributos específicos de produtos, processos e equipamentos de laboratório nos permitem modelar todo o processo de liofilização em ambientes de laboratório e manufatura. Esses atributos incluem uma ampla gama de fatores, incluindo formulação, temperaturas críticas, dimensões do frasco e volume de enchimento, pressão, transferência de calor e taxas de sublimação.
A modelagem também permite distinguir e compensar as variáveis de desempenho e design do equipamento, bem como o impacto dessas variáveis em formulações específicas e embalagens primárias. Isso produz uma linha de base informada antes de começar a colocar o produto na câmara de liofilização.
Disponibilidade de recursos e necessidade de modelagem de liofilização
Em uma economia de oferta e demanda, a escassez gera valor. Sujeitar o desenvolvimento do produto a abordagens de liofilização abaixo do ideal estende os prazos, adiciona custos evitáveis e atrasos em colocar a terapia nas mãos dos pacientes. Portanto, abordar metodicamente a liofilização com o parceiro de fabricação certo é fundamental para superar o desafio de desenvolver um processo de liofilização escalonável e repetível para substâncias biológicas medicamentosas.
É por isso que a modelagem, seguida por um exercício de liofilização em escala de bancada para confirmar os dados de modelagem em pequena escala, se mostra prudente como um primeiro passo crítico de pré-produção. Os benefícios para o desenvolvedor são que, ao adotar uma abordagem preventiva em escala de bancada, minimiza-se a substância medicamentosa e os custos associados necessários nos estágios iniciais. Então, uma vez que o produto avança para uma produção de GMP em maior escala, maior confiança é alcançada em torno dos parâmetros críticos do processo, levando a eficiências na utilização de substâncias medicamentosas, capacidade de fabricação otimizada e alocação apropriada de recursos de mão de obra de apoio.
Mitigando o risco de falha de fabricação de GMP
Há pressão para avançar rapidamente pelos estágios de desenvolvimento terapêutico para permitir a velocidade de comercialização. Uma compreensão completa dos requisitos no estágio de GMP é necessária para desenvolver um caminho robusto para a fabricação, que inclui um plano de mitigação, especialmente para tarefas identificadas no caminho crítico. Uma dessas considerações pode ser o momento, a disponibilidade e a quantidade do fármaco a granel para execuções de produção de liofilização em maior escala. Recomenda-se iniciar um ciclo em escala usando um substituto adequado para demonstrar a equivalência à substância medicamentosa real. Ao empregar um substituto, pode auxiliar na identificação de problemas de integridade física do produto, como nebulização nos frascos antes de utilizar a substância medicamentosa a granel potencialmente escassa e cara.
A modelagem e o trabalho em escala de bancada demonstram o impacto da transferência da fabricação em escala piloto para comercial, permitindo a caracterização completa do processo e do equipamento, revelando atributos de desempenho antes da liofilização da substância ativa final do medicamento em quantidades em escala de produção.
Reduza os custos de fabricação por meio de ciclos de liofilização minimizados
Novamente, com quantidades potencialmente limitadas de substância medicamentosa cara, alguns fabricantes escolhem abordagens conservadoras e mais longas para o tempo de ciclo de liofilização no estágio de desenvolvimento. Tempos de ciclo de liofilização mais longos resultam em custos adicionais e na redução da capacidade disponível.
O exercício de modelagem e escala de bancada cria uma oportunidade para otimizar a velocidade do ciclo de liofilização, determinando a taxa de liofilização mais rápida que uma formulação irá tolerar com segurança e eficácia, sem causar impacto negativo na aparência do bolo, estabilidade e outras características do produto. Esse conhecimento antecipado acelera os tempos de ciclo por meio da fabricação em escala subsequente, normalmente execuções clínicas e comerciais de fase final, enquanto reduz ainda mais o risco de falha ao verificar os parâmetros de liofilização desde o início.
Linha do tempo acelerada para status de FDA Fast-Track e Breakthrough
O FDA está concedendo status de terapia acelerada e inovadora em níveis recordes. Em 2018, 29 solicitações de designação fast-track foram concedidas e um recorde de 59 solicitações de designação de avanço foram aprovadas, contra 50, 46 e 32 nos três anos anteriores, respectivamente. Em março de 2019, 22 solicitações de designação de avanço já foram aprovadas, colocando o ano fiscal de 2019 em ritmo para superar o recorde do ano passado.
Com essas designações, vem uma maior urgência e um cronograma reduzido para prontidão comercial. Essa urgência pode acelerar a necessidade de coleta de dados, cronogramas de produção clínica e prontidão do CMC.
As designações aceleradas e inovadoras tornam os ganhos de eficiência e os benefícios de mitigação de risco da modelagem e da liofilização em escala de bancada ainda mais relevantes para os desenvolvedores. Eles fornecem uma caracterização do processo de linha de base que inspira confiança na probabilidade de acertar na primeira vez, o que, por sua vez, cria uma base para atender aos prazos clínicos e de comercialização acelerados.
A modelagem por computador e a liofilização em escala de bancada terão sucesso?
Apesar do resultado demonstrável de fornecer uma linha de base de pré-produção que mitiga riscos, economiza dinheiro e acelera o tempo de lançamento no mercado, a modelagem e a liofilização em escala de bancada não pegaram em todo o setor. Na verdade, uma pesquisa recente conduzida pelo fluxo de trabalho de liofilização do BioPhorum Operations Group (BPOG) (do qual AbbVie é membro) descobriu que poucos desenvolvedores e fabricantes de medicamentos estão implementando modelos para aumento de escala e transferência. Recentemente, em 2015, menos da metade dos principais fabricantes farmacêuticos pesquisados estavam aproveitando a modelagem para otimizar algum nível do processo de liofilização: desenvolvimento de processo (5 de 11), aumento de escala e transferência (5 de 11), otimização de processo ( 5 de 11), análise de desvios e tomada de decisão (2 de 11), e fornecer feedback adequado às autoridades regulatórias (1 de 11).
O BPOG tem trabalhado de forma colaborativa para mudar isso, definir as melhores práticas e mapear alguns padrões na esperança de promover uma adoção mais ampla da modelagem e da liofilização em escala de bancada.
Implementando modelagem e liofilização em escala de bancada em seu processo de desenvolvimento
A modelagem por computador e a liofilização em escala de bancada não dependem de nenhuma ciência ou tecnologia proprietária. Então, por que não é o padrão para desenvolvedores de drogas, particularmente aqueles que trabalham com substâncias biológicas de baixa quantidade? Resumindo, porque adiciona um nível de complexidade e tempo no front-end do desenvolvimento do produto. Apesar dos benefícios significativos de tempo, dinheiro e redução de risco obtidos posteriormente no processo, é um desafio para muitos desacelerar o ritmo de desenvolvimento. Se incorporado, o resultado da modelagem de transferência de calor pode fornecer uma compreensão de como a formulação influencia a resistência da torta, o liofilizador e a interação da substância medicamentosa, o impacto da escala do equipamento e o tipo para conduzir um processo otimizado.
O risco e o potencial de atraso do programa superam os riscos na fabricação robusta de longo prazo? Em operações, é altamente preferível que a fabricação não esteja no caminho crítico para a comercialização; ainda assim, encontrar o equilíbrio adequado entre velocidade e tempo de comercialização é uma decisão que é melhor tomada programa a programa. Uma entrada para essa decisão é avaliar o investimento relativamente pequeno de tempo e esforço de modelagem inicial, para gerar o melhor retorno de aprovação regulatória oportuna e robustez comercial.
Os produtores de medicamentos que consideram contratar um fabricante terceirizado para a fabricação e preenchimento / acabamento devem contemplar os benefícios da modelagem por computador e da liofilização em escala de bancada. Normalmente, o melhor momento para explorar esses benefícios é durante ou imediatamente após os ensaios clínicos da Fase 1, para garantir que o CMO possa se envolver totalmente e se beneficiar dos dados da Fase 1. Isso também é vantajoso para o desenvolvedor porque estudos essenciais fora do site comercial resultam na melhor chance de passar pelo desenvolvimento e mais perto de uma fase comercial no início de seu teste.