Acelere o desenvolvimento de moléculas complexas otimizando a síntese e a formulação químicas
Por Andreas Stolle, vice-presidente de Serviços de Desenvolvimento de Processos, API, e Peter Poechlauer, Ph.D., Gerente de Inovação, API, Thermo Fisher Scientific

As inovações em ciência e tecnologia nas últimas décadas permitem que os cientistas criem produtos farmacêuticos muito mais avançados para a indústria atual. Como resultado, os pacientes esperam legitimamente a medicação com menos efeitos colaterais e os médicos antecipam novas e melhores curas para doenças anteriormente intratáveis. Essas expectativas exigem que novos produtos farmacêuticos ofereçam uma vantagem sobre as terapias existentes, que as empresas patrocinadoras devem poder provar em ensaios clínicos. No entanto, eles também aumentam as dificuldades do desenvolvimento de medicamentos, devido à complexidade adicional do ingrediente farmacêutico ativo (API) e do sistema de administração necessário para esses medicamentos. Encontrar um equilíbrio entre uma API complexa, sua formulação e sua síntese requer equipamentos, conhecimentos e processos mais extensos do que aqueles normalmente necessários para o desenvolvimento tradicional de medicamentos.
Portanto, embora a complexidade dessas moléculas comece com sua química, qualquer empresa que entrar neste espaço provavelmente descobrirá que os desafios se estendem muito além disso. Não compreender e se preparar para eles no início do desenvolvimento pode resultar em gargalos que diminuem significativamente a produção e atrasam o tempo de comercialização de um medicamento.
Química e além: definindo a complexidade dos produtos farmacêuticos atuais
1. Química
Depois que um composto mostra eficácia, os cientistas devem apresentar uma rota de síntese aceitável. Isso é especialmente desafiador para os produtos farmacêuticos de hoje, devido à complexa estrutura química das novas APIs. Sua síntese inclui etapas mais químicas e intermediários potencialmente instáveis, reagentes perigosos e / ou condições de reação exigentes. Inicialmente, o foco de uma pequena empresa inovadora que busca uma dessas moléculas é estabelecer uma estrutura de chumbo em um organismo que produz um certo efeito farmacêutico. Eles fazem perguntas como: A estrutura do chumbo é absorvida e distribuída corretamente em um organismo? Como o metabolismo está funcionando? Como a excreção está funcionando?
Quando eles começam a otimizar os leads, eles criam um candidato a medicamentos com certas características estruturais e um padrão de substituição que alcança o efeito desejado. Até então, porém, todos os esforços foram feitos para descobrir como demonstrar eficácia. Os químicos provavelmente sintetizaram apenas pequenas quantidades do material (isto é, miligramas), provavelmente usando métodos elaborados para fazer isso. Pouca consideração foi colocada na formulação e ampliação, onde existem alguns dos maiores desafios e custos. Como os resultados clínicos do material produzido por uma rota anterior não podem ser aplicados perfeitamente ao material resultante de uma rota em larga escala, o resultado é um redesenho das sínteses químicas no final do processo. Em última análise, isso diminui o fornecimento das quantidades necessárias para os ensaios clínicos.
2. Propriedades das substâncias medicamentosas
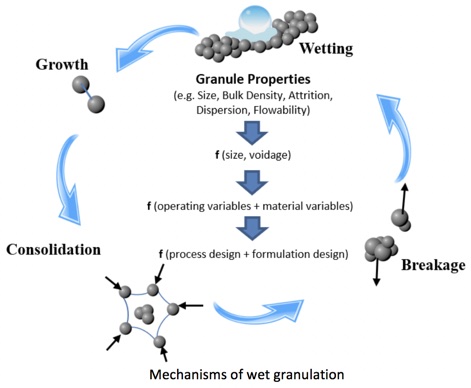
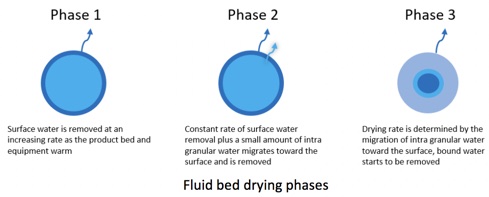
Como as drogas atuais são complexas, elas podem ter propriedades físicas exigentes e difíceis de controlar. Características físicas, como solubilidade e taxa de dissolução, devem ser determinadas muito cedo e em várias circunstâncias. As autoridades reguladoras também procurarão demonstrar que você controla os atributos desejados de qualidade química e física do produto recebidos pelo paciente. Além disso, as propriedades dessas novas APIs criam novos riscos nos procedimentos de manuseio, pois muitas têm "alta potência". Isso significa que eles são ativos em doses muito pequenas, portanto as pessoas que os fabricam precisam ser protegidas para evitar qualquer contato com a substância do medicamento. Uma potencial falta de estabilidade (ou seja, durabilidade) faz com que as APIs se degradem facilmente quando em contato com determinadas condições ambientais, como umidade e calor. Portanto, cuidados especiais devem ser tomados ao processá-los durante a formulação.
É por isso que, uma vez que um composto mostre eficácia, você deve concluir a pesquisa de rota para garantir que a síntese seja escalável em todas as fases do projeto. Isso pode ser desafiador, pois a base para um processo eficiente, escalonável e confiável é estabelecida em um momento em que não se sabe se o medicamento jamais verá a data de lançamento. Considerações como a sustentabilidade do medicamento também devem ser feitas nesta fase. Isso inclui uma análise de quão eficientemente as matérias-primas são usadas e se são ambientalmente aceitáveis (por exemplo, evite solventes halogenados, se possível).
Do lado da rentabilidade da sustentabilidade, você também deve garantir que o produto seja adequado para o futuro. Em outras palavras, como ela pode permanecer competitiva após o vencimento da patente e como pode ser feita para atender às metas de custo da saúde? Embora os custos da API durante os ensaios clínicos sejam extremamente altos, eles normalmente não são um foco, pois os volumes produzidos são muito pequenos. No entanto, assim que o produto é lançado, a empresa originadora terá que solicitar um preço elevado pelo medicamento para pagar por qualquer falha no desenvolvimento. O preço cai significativamente, no entanto, quando a droga se torna genérica e continua a cair até se tornar uma mercadoria, altura em que o preço é reduzido ainda mais. Ao analisar sua sustentabilidade antecipadamente, você pode estabelecer um processo que permita à sua empresa permanecer no negócio durante toda a vida útil do medicamento.
3. Necessidades de Formulação de Medicamentos As
novas APIs requerem conhecimento específico durante a formulação que pode ser usado para identificar e prevenir reações adversas que podem reduzir o prazo de validade de um medicamento posteriormente. Uma nova API pode não suportar as condições aplicadas em um processo de formulação convencional, e sua ação adequada em um paciente pode exigir sistemas de entrega de medicamentos específicos que direcionam uma API para o local pretendido, como um tecido ou tumor alvo.
Até o final da Fase II, a rota de síntese e formulação deve ser bloqueada e os custos da API precisam ser substancialmente reduzidos, pois esse será o processo usado para lançar o material. Todos os dados que seus cientistas criam nas fases 0-II não devem sofrer alterações durante o desenvolvimento; caso contrário, você pode entrar em uma situação em que certos estudos precisam ser repetidos. Por exemplo, alterações no polimorfo podem causar alterações na solubilidade e, portanto, na farmacocinética, o que pode forçar repetições de estudos. Uma maneira de gerenciar as complexidades dessas moléculas é ter várias e diferentes disciplinas envolvidas no desenvolvimento para garantir que os fatores mais críticos sejam levados em consideração desde o início.
Colaboração e comunicação: as chaves para o sucesso a longo prazo
Existem muitos tipos diferentes de conhecimentos envolvidos no desenvolvimento de medicamentos, e essas equipes de especialistas geralmente trabalham em silos. Além disso, muitas pequenas empresas que descobrem um novo conceito terapêutico não estão familiarizadas com os registros regulatórios, devido à sua experiência limitada com uma nova substância ativa. Os erros que podem ocorrer ao tentar levar sua descoberta do conceito ao suprimento comercial podem ser evitados combinando serviços de substâncias medicamentosas e desenvolvimento farmacêutico. Ao trabalhar juntos, uma equipe de formulação e os químicos podem colaborar e trocar conhecimento para desenvolver uma formulação que permita um fornecimento seguro de material durante os ensaios clínicos. As linhas de tempo só podem ser cumpridas se uma empresa empregar técnicas de ponta de fabricação, análise e formulação de API (por exemplo, síntese de fluxo contínuo ou técnicas de separação cromatográfica em larga escala). Isso requer equipes trabalhando em paralelo com muitas disciplinas diferentes, contribuindo com seus conhecimentos. Muitos desenvolvedores de novos medicamentos solicitam um processo de aprovação "acelerado", pois suas APIs podem responder a uma necessidade farmacêutica urgente (por exemplo, uma cura contra bactérias multirresistentes encontradas em hospitais). Um novo medicamento pode falhar a qualquer momento durante os ensaios clínicos. Portanto, os desenvolvedores de novos medicamentos, muitos dos quais são pequenas empresas, relutam em gastar muito tempo e dinheiro, enquanto o destino de seu candidato clínico ainda é incerto. Muitos desenvolvedores de novos medicamentos solicitam um processo de aprovação "acelerado", pois suas APIs podem responder a uma necessidade farmacêutica urgente (por exemplo, uma cura contra bactérias multirresistentes encontradas em hospitais). Um novo medicamento pode falhar a qualquer momento durante os ensaios clínicos. Portanto, os desenvolvedores de novos medicamentos, muitos dos quais são pequenas empresas, relutam em gastar muito tempo e dinheiro, enquanto o destino de seu candidato clínico ainda é incerto. Muitos desenvolvedores de novos medicamentos solicitam um processo de aprovação "acelerado", pois suas APIs podem responder a uma necessidade farmacêutica urgente (por exemplo, uma cura contra bactérias multirresistentes encontradas em hospitais). Um novo medicamento pode falhar a qualquer momento durante os ensaios clínicos. Portanto, os desenvolvedores de novos medicamentos, muitos dos quais são pequenas empresas, relutam em gastar muito tempo e dinheiro, enquanto o destino de seu candidato clínico ainda é incerto.
No entanto, os problemas de formulação dos produtos atualmente desenvolvidos estão surgindo com maior frequência e a complexidade pode comprometer o cronograma de desenvolvimento. Sua equipe deve aprender o máximo possível sobre uma API e um novo medicamento, independentemente da alta chance de falha nos estágios iniciais do desenvolvimento do medicamento. Deficiências no novo medicamento, como efeitos colaterais inesperados ou estabilidade insuficiente, podem se tornar visíveis bastante tarde no desenvolvimento. O candidato a fármaco então falha ou é necessário retrabalho caro ou repetição de ensaios clínicos. O tempo é especialmente precioso nesse estágio, pois a cada semana, mesmo dia, que um medicamento recém-lançado está sob proteção de patente, o remetente pode solicitar um preço muito mais alto (pagar pelo custo de desenvolvimento, incluindo falhas do candidato) .
Se uma empresa não procurar especialistas externos para ajudar a desenvolver sua molécula complexa, deve procurar recursos como as diretrizes do Conselho Internacional de Harmonização (ICH) para reconhecer e se preparar para os desafios relacionados. Também existem documentos técnicos do setor pelas autoridades reguladoras descrevendo e propondo maneiras de lidar com certos obstáculos. Webinars e conferências que incluam partes que compartilham sua experiência em lidar com esses problemas também devem ser considerados. No entanto, se uma empresa decidir se associar a um CDMO para o desenvolvimento de medicamentos, procure um que possua infraestrutura de ponta para realizar a fabricação de medicamentos e produtos farmacêuticos, bem como as competências necessárias para enfrentar esses desafios.
Publicado em 8/18 PATH0723