Engenharia de partículas para administração de medicamentos por inalação
Por Matthew Ferguson, Pesquisador Principal Sênior

INTRODUÇÃO
O desenvolvimento de terapias para doenças pulmonares continua a ser um mercado em expansão na pesquisa farmacêutica. As taxas de doenças pulmonares estão aumentando e as doenças respiratórias continuam a ser uma das principais causas de morte e incapacidade em todo o mundo. A amplitude de compostos biológicos e de pequenas moléculas que requerem administração pulmonar está se expandindo, aumentando a demanda por terapias de inalação que sejam eficazes, robustas, acessíveis e compatíveis para uso com inaladores de pó seco.
Um aspecto chave para o desenvolvimento bem-sucedido de terapias para atender à demanda do mercado é a fabricação de formulações com os atributos de qualidade do produto adequados para aplicação por inalação. Uma ampla gama de abordagens de engenharia de partículas está disponível, mas a seleção de qual tecnologia seria melhor para um determinado ingrediente farmacêutico ativo (API) pode ser um desafio, exigindo:
- estabelecer o perfil do produto alvo e o dispositivo de aplicação pretendido (por exemplo, inaladores de pó seco);
- avaliando propriedades de API e conduzindo experimentos de viabilidade focados; e
- avaliar os riscos associados a cada abordagem de engenharia de partículas candidata.
Ao compreender como as abordagens da engenharia de partículas influenciam os atributos do produto, os desenvolvedores podem estar na melhor posição para acelerar os testes clínicos, o desenvolvimento comercial e o tratamento do paciente.
Este resumo apresenta uma visão geral das tecnologias de engenharia de partículas e um estudo de caso no qual duas abordagens de engenharia são comparadas: secagem por spray (uma tecnologia "bottom-up") e moagem a jato (uma tecnologia "top-down"). Essas duas abordagens comprovadas e escalonáveis são relativamente simples e costumam ser um bom ponto de partida para determinar se uma tecnologia mais complexa é necessária para o sucesso.
ENGENHARIA DE PARTÍCULAS: MUITAS ESCOLHAS
Os avanços tecnológicos na engenharia de partículas tornam possível produzir partículas de drogas em dimensões de mícrons ou nanoescala usando muitos tipos de tecnologias. Geralmente, as tecnologias podem ser divididas nas seguintes classificações: (1) bottom-up, que envolve uma mudança de forma de solução para sólida; (2) de cima para baixo, que envolve a fresagem de um material até o tamanho desejado; e (3) abordagens de combinação, que envolvem o uso de várias tecnologias para lidar com situações complexas.
Tecnologias Bottom-Up
As tecnologias de baixo para cima envolvem a dissolução do API em solução a granel, formando gotículas e, em seguida, secando as gotículas para formar partículas. Essas tecnologias incluem secagem por spray, secagem por spray por congelamento, congelamento de película fina e tecnologias de fluido supercrítico. Muitas tecnologias de baixo para cima têm precedência de uso na indústria farmacêutica e são apoiadas por um amplo conhecimento das variáveis do processo. A secagem por spray é uma das tecnologias de baixo para cima mais comumente usadas devido à sua aplicabilidade a uma ampla gama de APIs e excipientes; a flexibilidade que oferece na engenharia da forma, tamanho e estado físico desejados das partículas (amorfo ou cristalino); confiabilidade; e escalabilidade.
Abordagens de cima para baixo
As tecnologias de cima para baixo envolvem moagem para reduzir o tamanho de partícula do API cristalino em um processo de uma ou várias etapas. O material de partida (ou seja, API) pode ser produzido usando muitos processos de fabricação diferentes, incluindo cristalização, secagem em massa ou liofilização, normalmente em um tamanho de partícula muito grande para ser respirável. A maioria das abordagens de cima para baixo são processos contínuos e escalonáveis que podem ser usados para micronizar muitos tipos de APIs. As abordagens de moagem são divididas entre moagem seca e úmida. Os tipos mais comuns de moagem a seco são moagem a jato, em que um fluxo de gás é usado para transportar partículas para fora da câmara de moagem, uma vez que o tamanho de partícula crítico é alcançado, e moagem de bolas. Na moagem úmida, estabilizadores podem ser adicionados para mitigar a energia de superfície mais alta das partículas e evitar problemas com propriedades de fluxo e uniformidade de conteúdo.
Abordagens de combinação
Combinações de tecnologias podem ser usadas em situações complexas, como quando um API tende a se aglomerar ou tem baixa solubilidade em solventes normalmente usados para secagem por pulverização. Um exemplo seria usar moagem para reduzir o tamanho de partícula, seguido por secagem por pulverização para revestir o núcleo API com uma camada fina de excipiente para alterar a química da superfície da formulação. Abordagens de combinação criam possibilidades de engenharia quase ilimitadas, mas suas vantagens podem ser compensadas pela complexidade e custo que adicionam ao trem do processo de manufatura.
CLASSIFICANDO AS ESCOLHAS
Embora todas as tecnologias descritas acima possam produzir pó com características ideais de administração por inalação, compreender as vantagens e limitações das várias opções pode economizar tempo e dinheiro de desenvolvimento valioso. Por exemplo, se a moagem e a secagem por pulverização produzirem características de partículas semelhantes, a moagem pode ser a escolha certa para um caminho de desenvolvimento mais simples e de alto rendimento.
Ao selecionar uma tecnologia, os desenvolvedores devem desenvolver uma compreensão completa das características físicas e químicas do API, incluindo quaisquer limitações, como sensibilidade à temperatura ou tendências de aglomeração, uma vez que estas podem excluir certas abordagens. Por exemplo, a moagem a jato funciona bem quando um API pode ser cristalizado de forma reproduzível com uma temperatura de fusão relativamente alta. No entanto, se a cristalinidade inicial ou o tamanho do cristalito do API variar, então o tamanho de partícula resultante do API moído pode ser mais dependente dos atributos do material de partida do que dos parâmetros do processo de moagem, apresentando aumento de escala e / ou lote para - desafios de variabilidade de lote. Da mesma forma, se a temperatura de fusão do API for muito baixa, aumenta a probabilidade de produção de material amorfo durante a moagem.
Se, depois de comparar as tecnologias candidatas, nenhuma escolha clara surgir, várias opções de tecnologia podem ser consideradas simultaneamente no início do desenvolvimento, auxiliadas por testes de viabilidade focados para examinar mais detalhadamente as escolhas. A avaliação simultânea é preferível a investigar tecnologias sequencialmente, uma vez que acelera o processo de desenvolvimento.
COMPARAÇÃO DE ESTUDO DE CASO
Como um caso de teste para comparar as abordagens de baixo para cima e de cima para baixo para a administração de drogas por inalação, o manitol cristalino foi projetado usando secagem por pulverização e moagem a jato para produzir partículas de tamanho respirável. O manitol tem sido usado em um teste de bronquite osmótico indireto e para aumentar a depuração mucociliar e tosse das secreções retidas no pulmão. Para a abordagem de baixo para cima, o manitol foi dissolvido em água e seco por pulverização usando parâmetros de processo selecionados para produzir a distribuição de tamanho de partícula desejada para administração de droga por inalação. Para a abordagem de cima para baixo, o mesmo material de partida foi moído a jato - novamente, com parâmetros especificamente escolhidos para produzir a distribuição correta do tamanho de partícula para a entrega do medicamento por inalação.
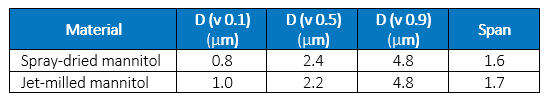
A Figura 1 mostra a distribuição de tamanho ponderado por volume das partículas e a Tabela 1 mostra a distribuição de tamanho de partícula. Como esses resultados mostram, o tamanho da partícula moída a jato era principalmente menor e tinha uma distribuição de tamanho mais ampla do que o material seco por pulverização. No entanto, as morfologias das partículas eram drasticamente diferentes, como mostrado nas imagens de microscopia eletrônica de varredura (MEV) na Figura 2. As partículas secas por spray eram esféricas, enquanto as partículas moídas a jato tinham bordas claramente definidas e seções transversais aproximadamente retangulares. Essas diferenças podem resultar em diferenças no tamanho aerodinâmico, mesmo que a densidade das partículas seja a mesma.
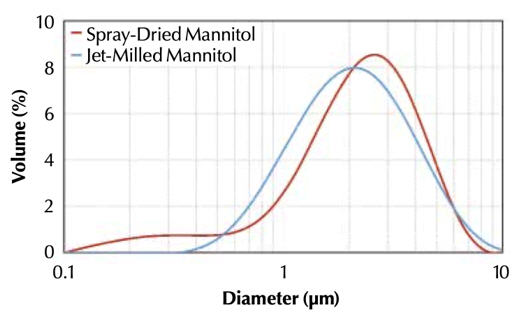
Figura 1: Distribuição geométrica do tamanho de partícula de manitol seco por pulverização e moído a jato, medido por difração a laser
Tabela 1: Distribuição de tamanho de partícula de manitol seco por pulverização e moído a jato, medido por difração a laser [D (v 0,1) = tamanho do percentil do 10º volume, D (v 0,5 = tamanho do percentil do volume 50, etc.
 / p>
/ p>

Figura 2: Imagens SEM de Manitol Spray-Dried (A) e Jet-Milled
Para avaliar como esses pós se comportariam na administração de drogas por inalação, um medidor aerodinâmico de partículas TSI (St. Paul, Minnesota, EUA) foi usado com análise de tempo de voo para determinar a distribuição aerodinâmica do tamanho das partículas dos pós. A Tabela 2 resume os dados, mostrando que o diâmetro aerodinâmico mediano de massa (MMAD) dos dois materiais foi semelhante. No entanto, a distribuição mais ampla das partículas grandes no manitol moído a jato resulta em uma fração menor sendo eficaz para entrega por inalação (definida como partículas com um diâmetro aerodinâmico inferior a 5 µm).
Tabela 2: Resumo do tamanho aerodinâmico, conforme medido pelo tempo de voo
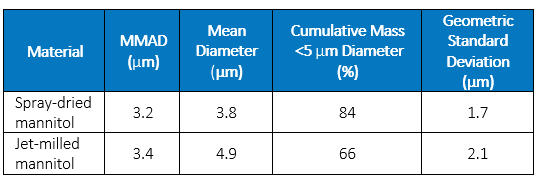
Nesse caso, tanto a secagem por spray quanto a moagem a jato parecem ser opções de tecnologia viáveis, apesar da diferença na morfologia das partículas. A comparação da cristalinidade entre os dois tipos de partículas pode ajudar a elucidar se qualquer um dos materiais contém material amorfo significativo ou se a estrutura do cristal pode ser suscetível a alterações com o calor e a umidade ao longo do tempo.
RESUMO
A seleção da melhor tecnologia para um produto de entrega de medicamento por inalação requer uma consideração cuidadosa das características do API e do perfil do produto alvo. Geralmente, tecnologias simples, escalonáveis e bem caracterizadas são preferíveis, tornando a secagem por spray e moagem a jato excelentes primeiras escolhas para muitos APIs. Avaliando essas abordagens inicialmente, os desenvolvedores podem determinar se uma tecnologia mais complicada é necessária e qual caminho é mais adequado para a API e o produto em questão. Se várias tecnologias forem adequadas, os desenvolvedores podem otimizar a velocidade para a clínica e o paciente, levando em consideração o rendimento do processo, o rendimento e a disponibilidade do equipamento.
SOBRE LONZA
A engenharia de partículas - crítica para alcançar a distribuição de tamanho de partícula necessária para a entrega eficaz de medicamentos por inalação - é um ponto forte da Lonza. As décadas de experiência da empresa, especialização em formulação e engenharia e ampla gama de recursos de fabricação tornam a Lonza a parceira preferida para aplicações de entrega de medicamentos por inalação. O desenvolvimento da empresa e as capacidades de fabricação de pó para inalação clínica - localizadas em Bend, Oregon, EUA - dão suporte a todas as fases de desenvolvimento do produto para inalação. Secagem por pulverização em pequena escala, moagem úmida e moagem a jato estão disponíveis para o trabalho inicial de viabilidade. Salas limpas de última geração para secagem por spray e enchimento de cápsulas também estão disponíveis, assim como uma suíte de alta contenção para o manuseio seguro de compostos biológicos e pequenos de alta potência. A Lonza possui recursos de moagem a jato e secagem por spray em escala clínica e comercial em locais em todo o mundo. Para aprender mais, por favorvisite nosso website .








