INTRODUÇÃO AOS ESTUDOS DE DEGRADAÇÃO FORÇADA DE SUBSTÂNCIAS E PRODUTOS FARMACÊUTICOS
- 01/09/2020
- Publicado por: LSTI-Editor
-
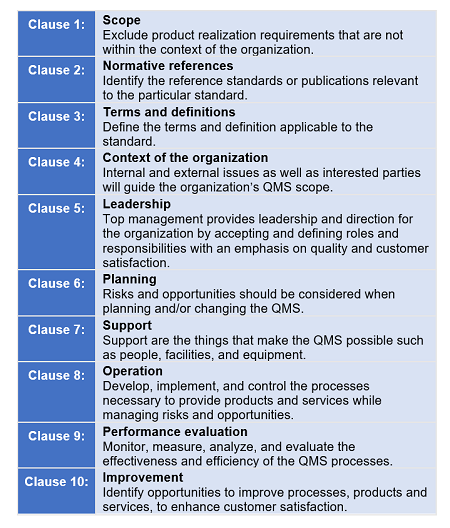
O estudo de degradação forçada é considerado um aspecto analítico vital do programa de desenvolvimento de medicamentos para pequenas moléculas. A degradação forçada, comumente conhecida como teste de estresse, é realizada para demonstrar como especificidade o desenvolvimento de um método analítico de indicação de estabilidade, utilizando cromatografia líquida de alta eficiência (HPLC), ou seja, um método analítico único capaz de separar os picos de degradantes de o pico da substância / medicamento. De acordo com as diretrizes da Conferência Internacional de Harmonização (ICH) (Q1A), estudos de estabilidade precisam ser realizados para propor o prazo de validade de novas substâncias e / ou medicamentos. Os estudos de vida de prateleira fazem parte de vários envios regulatórios ao FDA.
Geralmente, três tipos de estudos de estabilidade precisam ser realizados para propor a vida útil de uma substância e / ou medicamento: estabilidade acelerada (ACC), estabilidade intermediária (INS) e estabilidade da temperatura ambiente controlada (CRT). A duração do estudo acelerado é de cerca de seis meses, os estudos de estabilidade intermediária e estabilidade à temperatura ambiente levam cerca de 12 a 24 meses. Um estudo de estabilidade é realizado para determinar a estabilidade intrínseca da molécula e, durante o estudo, espera-se que a substância / medicamento se degrade / se decomponha e gere outras moléculas, conhecidas como impurezas. Durante os estudos de degradação forçada, várias condições de tensão são deliberadamente aplicadas para degradar / decompor o composto principal e gerar impurezas, que deve separar do composto principal e um do outro. Portanto, os estudos de degradação forçada são uma ferramenta para estimar as impurezas degradantes / decompostas que ocorreriam durante os estudos de estabilidade e são usadas para propor o prazo de validade das novas substâncias e / ou medicamentos.
Várias técnicas / equipamentos analíticos podem ser usados para separar e estimar todos os compostos degradantes que se espera estarem presentes no momento do estudo de degradação forçada. HPLC com detector UV (HPLC-UV) e detector de matriz de fotodiodo (HPLC-PDA) são técnicas bem conhecidas e são amplamente utilizadas nas indústrias farmacêuticas durante estudos de degradação forçada, como parte do desenvolvimento e validação do método de indicação de estabilidade. HPLC com detector de massa (LC-MS), cromatografia gasosa com detector de massa (GC-MS) e espectroscopia de ressonância magnética nuclear (RMN) são técnicas importantes para identificar a estrutura dos degradantes.
- 01/09/2020
- Publicado por: LSTI-Editor
O estudo de degradação forçada é considerado um aspecto analítico vital do programa de desenvolvimento de medicamentos para pequenas moléculas. A degradação forçada, comumente conhecida como teste de estresse, é realizada para demonstrar como especificidade o desenvolvimento de um método analítico de indicação de estabilidade, utilizando cromatografia líquida de alta eficiência (HPLC), ou seja, um método analítico único capaz de separar os picos de degradantes de o pico da substância / medicamento. De acordo com as diretrizes da Conferência Internacional de Harmonização (ICH) (Q1A), estudos de estabilidade precisam ser realizados para propor o prazo de validade de novas substâncias e / ou medicamentos. Os estudos de vida de prateleira fazem parte de vários envios regulatórios ao FDA.
Geralmente, três tipos de estudos de estabilidade precisam ser realizados para propor a vida útil de uma substância e / ou medicamento: estabilidade acelerada (ACC), estabilidade intermediária (INS) e estabilidade da temperatura ambiente controlada (CRT). A duração do estudo acelerado é de cerca de seis meses, os estudos de estabilidade intermediária e estabilidade à temperatura ambiente levam cerca de 12 a 24 meses. Um estudo de estabilidade é realizado para determinar a estabilidade intrínseca da molécula e, durante o estudo, espera-se que a substância / medicamento se degrade / se decomponha e gere outras moléculas, conhecidas como impurezas. Durante os estudos de degradação forçada, várias condições de tensão são deliberadamente aplicadas para degradar / decompor o composto principal e gerar impurezas, que deve separar do composto principal e um do outro. Portanto, os estudos de degradação forçada são uma ferramenta para estimar as impurezas degradantes / decompostas que ocorreriam durante os estudos de estabilidade e são usadas para propor o prazo de validade das novas substâncias e / ou medicamentos.
Várias técnicas / equipamentos analíticos podem ser usados para separar e estimar todos os compostos degradantes que se espera estarem presentes no momento do estudo de degradação forçada. HPLC com detector UV (HPLC-UV) e detector de matriz de fotodiodo (HPLC-PDA) são técnicas bem conhecidas e são amplamente utilizadas nas indústrias farmacêuticas durante estudos de degradação forçada, como parte do desenvolvimento e validação do método de indicação de estabilidade. HPLC com detector de massa (LC-MS), cromatografia gasosa com detector de massa (GC-MS) e espectroscopia de ressonância magnética nuclear (RMN) são técnicas importantes para identificar a estrutura dos degradantes.
Aplicações Importantes do Estudo de Degradação Forçada
A degradação forçada é um estudo analítico crítico para o desenvolvimento de métodos indicadores de estabilidade a serem usados por empresas farmacêuticas como parte de envios regulatórios ao FDA. Algumas das aplicações dos estudos são:
- Desenvolver e validar métodos de indicação de estabilidade, de acordo com as diretrizes da ICH.
- Identificar estrutura e toxicidade e definir especificações de degradantes ou impurezas.
- Propor o prazo de validade do produto sem informações de estabilidade em tempo real.
- Para otimizar formulações e selecionar placebos para medicamentos para evitar interferências.
- Justificar impurezas relacionadas ao processo ou produtos de degradação.
- Apoiar a identificação da causa raiz durante investigações fora de especificação (OOS) / laboratório.
- Acompanhar o arquivo mestre de medicamentos e as submissões da ANDA / NDA e IND à FDA.
A degradação forçada é um estudo analítico crítico para o desenvolvimento de métodos indicadores de estabilidade a serem usados por empresas farmacêuticas como parte de envios regulatórios ao FDA. Algumas das aplicações dos estudos são:
- Desenvolver e validar métodos de indicação de estabilidade, de acordo com as diretrizes da ICH.
- Identificar estrutura e toxicidade e definir especificações de degradantes ou impurezas.
- Propor o prazo de validade do produto sem informações de estabilidade em tempo real.
- Para otimizar formulações e selecionar placebos para medicamentos para evitar interferências.
- Justificar impurezas relacionadas ao processo ou produtos de degradação.
- Apoiar a identificação da causa raiz durante investigações fora de especificação (OOS) / laboratório.
- Acompanhar o arquivo mestre de medicamentos e as submissões da ANDA / NDA e IND à FDA.
Seleção e procedimentos da condição de degradação forçada
De acordo com as diretrizes da ICH e as práticas comuns da indústria, a degradação forçada geralmente é realizada em diferentes condições de estresse, como ácido, álcalis, peróxido, térmica e UV, juntamente com uma amostra de controle. Não há diretrizes industriais sobre quanta degradação deve ser alcançada; no entanto, pelas práticas industriais atuais, deve-se obter uma degradação de 5 a 30% em qualquer uma das condições de estresse aplicadas. O objetivo da degradação a ser alcançada através do teste de estresse é imitar as condições de estabilidade da temperatura da sala de controle. Nos casos em que são observadas degradações maiores ou menores, as condições ou concentrações do reagente devem ser otimizadas. O balanço de massa deve ser demonstrado durante o estudo de degradação e deve ser em torno de 100%, levando em consideração as margens de erros analíticos.
Durante o estudo de degradação forçada, qualquer lote que não faça parte da submissão regulatória pode ser usado. No caso de um medicamento, se vários pontos fortes estiverem disponíveis com os mesmos placebos e quantidades diferentes, a força que tiver a maior proporção de placebo versus ingrediente farmacêutico ativo (API) deve ser usada. Onde os placebos são diferentes, deve-se demonstrar a degradação forçada de todos os pontos fortes. Durante o estudo de degradação do medicamento, o placebo e a API devem ser demonstrados para identificar as vias de degradação reais. Onde os placebos são diferentes para diferentes pontos fortes do medicamento, todos os placebos devem ser considerados para o estudo de degradação.
A seguir (Tabela 1) estão as condições de degradação sugeridas conforme as práticas industriais atuais e aceitas pelo FDA durante os envios de DMF / ANDA / NDA e IND para aprovação regulatória.
Tabela 1: Condições experimentais sugeridas para estudos de degradação forçada
 A degradação deve ser realizada na forma sólida ou em solução, mas recomenda-se que seja realizada na forma de solução usando a fase diluente / móvel para obter um efeito homogêneo. Os estudos de degradação podem ser iniciados com condições adversas (ou seja, alta concentração de reagente com alta temperatura) para reduzir o tempo do estudo. Nos casos em que é encontrada degradação superior a 30%, condições mais brandas devem ser aplicadas, reduzindo a concentração do reagente, diminuindo a temperatura etc. As condições de degradação devem ser otimizadas para atingir um objetivo com base no resultado inicial da degradação. O pH deve ser ajustado para cerca de 7,0 para degradação ácida e alcalina para prolongar a vida útil da coluna cromatográfica. Se a degradação não for encontrada em nenhuma das condições acima, diferentes reagentes e condições podem ser aplicados, por exemplo, H2SO4, Zn, etc. Existem algumas moléculas que não se degradam sob condições adversas e, portanto, são consideradas moléculas estáveis nas rochas. Esse tipo de molécula não irá gerar impurezas adicionais / picos degradantes durante um estudo de estabilidade.
A degradação deve ser realizada na forma sólida ou em solução, mas recomenda-se que seja realizada na forma de solução usando a fase diluente / móvel para obter um efeito homogêneo. Os estudos de degradação podem ser iniciados com condições adversas (ou seja, alta concentração de reagente com alta temperatura) para reduzir o tempo do estudo. Nos casos em que é encontrada degradação superior a 30%, condições mais brandas devem ser aplicadas, reduzindo a concentração do reagente, diminuindo a temperatura etc. As condições de degradação devem ser otimizadas para atingir um objetivo com base no resultado inicial da degradação. O pH deve ser ajustado para cerca de 7,0 para degradação ácida e alcalina para prolongar a vida útil da coluna cromatográfica. Se a degradação não for encontrada em nenhuma das condições acima, diferentes reagentes e condições podem ser aplicados, por exemplo, H2SO4, Zn, etc. Existem algumas moléculas que não se degradam sob condições adversas e, portanto, são consideradas moléculas estáveis nas rochas. Esse tipo de molécula não irá gerar impurezas adicionais / picos degradantes durante um estudo de estabilidade.
De acordo com as diretrizes da ICH e as práticas comuns da indústria, a degradação forçada geralmente é realizada em diferentes condições de estresse, como ácido, álcalis, peróxido, térmica e UV, juntamente com uma amostra de controle. Não há diretrizes industriais sobre quanta degradação deve ser alcançada; no entanto, pelas práticas industriais atuais, deve-se obter uma degradação de 5 a 30% em qualquer uma das condições de estresse aplicadas. O objetivo da degradação a ser alcançada através do teste de estresse é imitar as condições de estabilidade da temperatura da sala de controle. Nos casos em que são observadas degradações maiores ou menores, as condições ou concentrações do reagente devem ser otimizadas. O balanço de massa deve ser demonstrado durante o estudo de degradação e deve ser em torno de 100%, levando em consideração as margens de erros analíticos.
Durante o estudo de degradação forçada, qualquer lote que não faça parte da submissão regulatória pode ser usado. No caso de um medicamento, se vários pontos fortes estiverem disponíveis com os mesmos placebos e quantidades diferentes, a força que tiver a maior proporção de placebo versus ingrediente farmacêutico ativo (API) deve ser usada. Onde os placebos são diferentes, deve-se demonstrar a degradação forçada de todos os pontos fortes. Durante o estudo de degradação do medicamento, o placebo e a API devem ser demonstrados para identificar as vias de degradação reais. Onde os placebos são diferentes para diferentes pontos fortes do medicamento, todos os placebos devem ser considerados para o estudo de degradação.
A seguir (Tabela 1) estão as condições de degradação sugeridas conforme as práticas industriais atuais e aceitas pelo FDA durante os envios de DMF / ANDA / NDA e IND para aprovação regulatória.
Tabela 1: Condições experimentais sugeridas para estudos de degradação forçada
A degradação deve ser realizada na forma sólida ou em solução, mas recomenda-se que seja realizada na forma de solução usando a fase diluente / móvel para obter um efeito homogêneo. Os estudos de degradação podem ser iniciados com condições adversas (ou seja, alta concentração de reagente com alta temperatura) para reduzir o tempo do estudo. Nos casos em que é encontrada degradação superior a 30%, condições mais brandas devem ser aplicadas, reduzindo a concentração do reagente, diminuindo a temperatura etc. As condições de degradação devem ser otimizadas para atingir um objetivo com base no resultado inicial da degradação. O pH deve ser ajustado para cerca de 7,0 para degradação ácida e alcalina para prolongar a vida útil da coluna cromatográfica. Se a degradação não for encontrada em nenhuma das condições acima, diferentes reagentes e condições podem ser aplicados, por exemplo, H2SO4, Zn, etc. Existem algumas moléculas que não se degradam sob condições adversas e, portanto, são consideradas moléculas estáveis nas rochas. Esse tipo de molécula não irá gerar impurezas adicionais / picos degradantes durante um estudo de estabilidade.
Caracterizações e Balanço de Massa do Estudo de Degradação Forçada
Todas as soluções devem ser analisadas usando métodos analíticos adequados pré-desenvolvidos, juntamente com as soluções de diluente, placebo e amostra de controle. Os critérios de aceitação sugeridos durante a degradação forçada devem ser os seguintes:
- Degradação de pelo menos 5 a 30 por cento deve ser observada em qualquer uma das condições.
- O pico principal deve ser bem separado do diluente, placebo, picos conhecidos e picos degradantes gerados durante a degradação.
- O pico principal deve ser puro, ou seja, nenhum outro pico deve ser mesclado com o pico principal. Geralmente, pode ser determinado usando software como o Empower 3 se o limite de pureza for maior que o ângulo de pureza, o que significa que o pico é puro. O software avançado pode gerar uma imagem 3D de todos os picos de cada cromatograma, o que ajuda a identificar a pureza do pico, conforme mostrado na figura a seguir.
 Nota: É possível que o pico principal esteja separado de todos os picos e seja puro; no entanto, impurezas conhecidas ou picos degradantes gerados durante o estudo de degradação não são puros e podem ser mesclados com outros picos. Nesse caso, devem ser feitas tentativas para modificar o método analítico para separar todos os picos um do outro. Portanto, também é recomendado que a pureza do pico seja avaliada para todos os picos (pico principal, picos de impureza conhecidos e todos os picos degradantes) para evitar consequências durante o estudo de estabilidade. Quando as tentativas de obter a pureza máxima dos picos degradantes falham, os detalhes do desenvolvimento do método devem ser documentados e uma abordagem baseada em risco deve ser aplicada para aceitar o método como indicador de estabilidade.
Durante a degradação forçada, muitos picos mais degradantes podem ser gerados a partir do pico principal (ativo). A porcentagem de degradação do pico principal (ativo) deve ser calculada após a conclusão da análise. A fórmula a seguir é geralmente usada nas indústrias farmacêuticas e aceita pelo FDA para calcular a porcentagem de degradação.
Nota: É possível que o pico principal esteja separado de todos os picos e seja puro; no entanto, impurezas conhecidas ou picos degradantes gerados durante o estudo de degradação não são puros e podem ser mesclados com outros picos. Nesse caso, devem ser feitas tentativas para modificar o método analítico para separar todos os picos um do outro. Portanto, também é recomendado que a pureza do pico seja avaliada para todos os picos (pico principal, picos de impureza conhecidos e todos os picos degradantes) para evitar consequências durante o estudo de estabilidade. Quando as tentativas de obter a pureza máxima dos picos degradantes falham, os detalhes do desenvolvimento do método devem ser documentados e uma abordagem baseada em risco deve ser aplicada para aceitar o método como indicador de estabilidade.
Durante a degradação forçada, muitos picos mais degradantes podem ser gerados a partir do pico principal (ativo). A porcentagem de degradação do pico principal (ativo) deve ser calculada após a conclusão da análise. A fórmula a seguir é geralmente usada nas indústrias farmacêuticas e aceita pelo FDA para calcular a porcentagem de degradação.

Usando a porcentagem de degradação, o balanço de massa deve ser avaliado para todos os picos degradantes. O balanço de massa deve ser de cerca de 100%, considerando a margem de erros durante a análise.
HPLC com detector de massa (LC-MS e LC-MS / MS), cromatografia gasosa com detector de massa (GC-MS) e espectroscopia de ressonância magnética nuclear (RMN) são técnicas importantes para identificar a estrutura e o balanço de massa dos degradantes. A caracterização dos degradantes pode ser determinada usando técnicas analíticas avançadas como LC-RMN. Cromatografia em camada fina (TLC), cromatografia em coluna e HPLC preparativa podem ser usadas para isolar as impurezas do resíduo ou filtrado gerado durante o processo de fabricação da API.
Todas as soluções devem ser analisadas usando métodos analíticos adequados pré-desenvolvidos, juntamente com as soluções de diluente, placebo e amostra de controle. Os critérios de aceitação sugeridos durante a degradação forçada devem ser os seguintes:
- Degradação de pelo menos 5 a 30 por cento deve ser observada em qualquer uma das condições.
- O pico principal deve ser bem separado do diluente, placebo, picos conhecidos e picos degradantes gerados durante a degradação.
- O pico principal deve ser puro, ou seja, nenhum outro pico deve ser mesclado com o pico principal. Geralmente, pode ser determinado usando software como o Empower 3 se o limite de pureza for maior que o ângulo de pureza, o que significa que o pico é puro. O software avançado pode gerar uma imagem 3D de todos os picos de cada cromatograma, o que ajuda a identificar a pureza do pico, conforme mostrado na figura a seguir.
Nota: É possível que o pico principal esteja separado de todos os picos e seja puro; no entanto, impurezas conhecidas ou picos degradantes gerados durante o estudo de degradação não são puros e podem ser mesclados com outros picos. Nesse caso, devem ser feitas tentativas para modificar o método analítico para separar todos os picos um do outro. Portanto, também é recomendado que a pureza do pico seja avaliada para todos os picos (pico principal, picos de impureza conhecidos e todos os picos degradantes) para evitar consequências durante o estudo de estabilidade. Quando as tentativas de obter a pureza máxima dos picos degradantes falham, os detalhes do desenvolvimento do método devem ser documentados e uma abordagem baseada em risco deve ser aplicada para aceitar o método como indicador de estabilidade.
Durante a degradação forçada, muitos picos mais degradantes podem ser gerados a partir do pico principal (ativo). A porcentagem de degradação do pico principal (ativo) deve ser calculada após a conclusão da análise. A fórmula a seguir é geralmente usada nas indústrias farmacêuticas e aceita pelo FDA para calcular a porcentagem de degradação.
Usando a porcentagem de degradação, o balanço de massa deve ser avaliado para todos os picos degradantes. O balanço de massa deve ser de cerca de 100%, considerando a margem de erros durante a análise.
HPLC com detector de massa (LC-MS e LC-MS / MS), cromatografia gasosa com detector de massa (GC-MS) e espectroscopia de ressonância magnética nuclear (RMN) são técnicas importantes para identificar a estrutura e o balanço de massa dos degradantes. A caracterização dos degradantes pode ser determinada usando técnicas analíticas avançadas como LC-RMN. Cromatografia em camada fina (TLC), cromatografia em coluna e HPLC preparativa podem ser usadas para isolar as impurezas do resíduo ou filtrado gerado durante o processo de fabricação da API.
Você precisa entender o processo de desenvolvimento de novos medicamentos?
Confira webinar de Albert Yehaskel, “ Desenvolvimento de Medicamentos 101 - como a droga é feita ”
Procurando uma visão geral de alto nível de como um medicamento é produzido?
Confira webinar de Albert Yehaskel, “ Desenvolvimento de Medicamentos 101 - como a droga é feita ”
Confira webinar de Albert Yehaskel, “ Desenvolvimento de Medicamentos 101 - como a droga é feita ”
Desenvolvimento de Métodos Indicadores de Estabilidade Incorporando Estudo de Degradação de Força
Durante o estudo de degradação forçada, podem ser gerados produtos de degradação que podem ser mais do que o esperado durante o ACC. Amostras degradadas, juntamente com impurezas conhecidas, devem ser usadas para desenvolver o método analítico. Essas amostras representam o pior cenário e todos os picos (pico principal, picos degradados e impurezas conhecidas) devem ser bem separados um do outro. Nos casos em que o método analítico não é capaz de separar todos os picos, o método precisa ser reconstruído para obter a separação de todos os picos. A referência a livros, literatura e experiência é um fator-chave no desenvolvimento do método analítico. Um dos livros respeitáveis é o Desenvolvimento prático do método HPLCde R. Snyder. Isso ajudará os usuários a entender como obter uma melhor separação de todos os picos. Uma vez desenvolvido um método analítico capaz de separar todos os picos das amostras degradadas, significa que esse método é capaz de analisar todas as amostras de estabilidade e, portanto, esse método analítico será considerado indicador de estabilidade. A validação do método deve ser realizada para demonstrar que o método analítico é adequado para a finalidade a que se destina (consulte Diretrizes da ICH Q2 (R1), Validação de procedimentos analíticos). As informações de degradação forçada também podem ajudar a determinar a causa raiz durante as investigações de OOS / laboratório durante a análise da amostra, por exemplo, se algum pico inesperado for observado durante a análise da amostra que não foi observado durante qualquer condição de degradação forçada,
Durante o estudo de degradação forçada, podem ser gerados produtos de degradação que podem ser mais do que o esperado durante o ACC. Amostras degradadas, juntamente com impurezas conhecidas, devem ser usadas para desenvolver o método analítico. Essas amostras representam o pior cenário e todos os picos (pico principal, picos degradados e impurezas conhecidas) devem ser bem separados um do outro. Nos casos em que o método analítico não é capaz de separar todos os picos, o método precisa ser reconstruído para obter a separação de todos os picos. A referência a livros, literatura e experiência é um fator-chave no desenvolvimento do método analítico. Um dos livros respeitáveis é o Desenvolvimento prático do método HPLCde R. Snyder. Isso ajudará os usuários a entender como obter uma melhor separação de todos os picos. Uma vez desenvolvido um método analítico capaz de separar todos os picos das amostras degradadas, significa que esse método é capaz de analisar todas as amostras de estabilidade e, portanto, esse método analítico será considerado indicador de estabilidade. A validação do método deve ser realizada para demonstrar que o método analítico é adequado para a finalidade a que se destina (consulte Diretrizes da ICH Q2 (R1), Validação de procedimentos analíticos). As informações de degradação forçada também podem ajudar a determinar a causa raiz durante as investigações de OOS / laboratório durante a análise da amostra, por exemplo, se algum pico inesperado for observado durante a análise da amostra que não foi observado durante qualquer condição de degradação forçada,
Conclusão
A degradação forçada é um estudo essencial que fornece conhecimento e julgamento para desenvolver um método analítico de indicação de estabilidade. Este estudo deve ser realizado e deve fazer parte de envios regulatórios. Este estudo também ajuda a estabelecer a especificação e o prazo de validade de uma substância ou medicamento. As informações derivadas do estudo ajudarão a melhorar a formulação, o processo de fabricação e as condições de armazenamento do produto. Além disso, o estudo de degradação forçada também ajuda a descobrir causas-raiz ou qualquer contaminação em potencial durante o processo de fabricação ou durante a análise laboratorial. Portanto, o estudo de degradação forçada deve ser demonstrado no momento do desenvolvimento do método e antes da submissão do dossiê regulatório ao FDA.
A degradação forçada é um estudo essencial que fornece conhecimento e julgamento para desenvolver um método analítico de indicação de estabilidade. Este estudo deve ser realizado e deve fazer parte de envios regulatórios. Este estudo também ajuda a estabelecer a especificação e o prazo de validade de uma substância ou medicamento. As informações derivadas do estudo ajudarão a melhorar a formulação, o processo de fabricação e as condições de armazenamento do produto. Além disso, o estudo de degradação forçada também ajuda a descobrir causas-raiz ou qualquer contaminação em potencial durante o processo de fabricação ou durante a análise laboratorial. Portanto, o estudo de degradação forçada deve ser demonstrado no momento do desenvolvimento do método e antes da submissão do dossiê regulatório ao FDA.
Referências:
- Teste de estabilidade ICH Q1A (R2) de novas substâncias e produtos para medicamentos https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances -products-step-5_en.pdf
- ICH Q2 (R1) Validação de procedimentos analíticos https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analytical-procedures-text-methodology-step-5_pt .pdf
- Impurezas ICH Q3A (R2) em novas substâncias medicamentosas https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-3-r2-impurities-new-drug-substances-step-5_en. pdf
- ICH Q3B (R2) Impurezas em novos medicamentos https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-3-b-r2-impurities-new-drug-products-step- 5_en.pdf
- Especificações da ICH Q6A: procedimentos de teste e critérios de aceitação para novas substâncias e novos medicamentos (substâncias químicas) https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-6-test-procedures -ac critérios de aceitação-novas-drogas-substâncias-novos-drogas-produtos-químicos_pt.pdf
- Capítulo Geral da USP <1225>: Validação de Procedimentos Compêndios
- Manual do Empower PDA Software - Guia de introdução
- Snyder, L., Kirkland, J. e Glajch, J. Practical HPLC Method Development . Wiley, 1997.
- Guia para Iniciantes em Cromatografia Líquida (Série Waters), 1ª Edição. Waters Corporation, 2014.
- Ahuja, S. e Rasmussen, H. HPLC Method Development for Pharmaceuticals. Academic Press, 2007.
- Kats, R. “Estudos de degradação forçada: considerações regulatórias e implementação.” BioPharm International , 01 de julho de 2005.
- Reynolds D., et al. “Orientação disponível e melhores práticas para a realização de estudos de degradação forçada.” Pharmaceutical Technology, 1 de fevereiro de 2002.
- Teste de estabilidade ICH Q1A (R2) de novas substâncias e produtos para medicamentos https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances -products-step-5_en.pdf
- ICH Q2 (R1) Validação de procedimentos analíticos https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analytical-procedures-text-methodology-step-5_pt .pdf
- Impurezas ICH Q3A (R2) em novas substâncias medicamentosas https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-3-r2-impurities-new-drug-substances-step-5_en. pdf
- ICH Q3B (R2) Impurezas em novos medicamentos https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-3-b-r2-impurities-new-drug-products-step- 5_en.pdf
- Especificações da ICH Q6A: procedimentos de teste e critérios de aceitação para novas substâncias e novos medicamentos (substâncias químicas) https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-6-test-procedures -ac critérios de aceitação-novas-drogas-substâncias-novos-drogas-produtos-químicos_pt.pdf
- Capítulo Geral da USP <1225>: Validação de Procedimentos Compêndios
- Manual do Empower PDA Software - Guia de introdução
- Snyder, L., Kirkland, J. e Glajch, J. Practical HPLC Method Development . Wiley, 1997.
- Guia para Iniciantes em Cromatografia Líquida (Série Waters), 1ª Edição. Waters Corporation, 2014.
- Ahuja, S. e Rasmussen, H. HPLC Method Development for Pharmaceuticals. Academic Press, 2007.
- Kats, R. “Estudos de degradação forçada: considerações regulatórias e implementação.” BioPharm International , 01 de julho de 2005.
- Reynolds D., et al. “Orientação disponível e melhores práticas para a realização de estudos de degradação forçada.” Pharmaceutical Technology, 1 de fevereiro de 2002.
Sobre o autor:
Vinubhai Patolia tem 20 anos de experiência em pesquisa e desenvolvimento analítico e controle de qualidade na indústria farmacêutica. Após concluir seu mestrado (química orgânica), trabalhou no Zydus Cadila, no Centro de Pesquisa Avançada Farmacêutica Sun e na Actavis Pharma. Mais recentemente, ele foi gerente de pesquisa e desenvolvimento analítico da Sun Pharma em Cranbury, NJ. Durante sua carreira, Patolia adquiriu uma experiência significativa no desenvolvimento de métodos, validação de métodos, transferência de métodos e resposta a deficiências do FDA em relação a substâncias, medicamentos e excipientes para submissões de ANDA / NDA. Ele trabalhou em várias formas de dosagem, incluindo dose oral sólida, tópica, suspensão e injetável.
Vinubhai Patolia tem 20 anos de experiência em pesquisa e desenvolvimento analítico e controle de qualidade na indústria farmacêutica. Após concluir seu mestrado (química orgânica), trabalhou no Zydus Cadila, no Centro de Pesquisa Avançada Farmacêutica Sun e na Actavis Pharma. Mais recentemente, ele foi gerente de pesquisa e desenvolvimento analítico da Sun Pharma em Cranbury, NJ. Durante sua carreira, Patolia adquiriu uma experiência significativa no desenvolvimento de métodos, validação de métodos, transferência de métodos e resposta a deficiências do FDA em relação a substâncias, medicamentos e excipientes para submissões de ANDA / NDA. Ele trabalhou em várias formas de dosagem, incluindo dose oral sólida, tópica, suspensão e injetável.