O que são adoçantes?
A importância dos Adoçantes na Industria Farmacêutica.
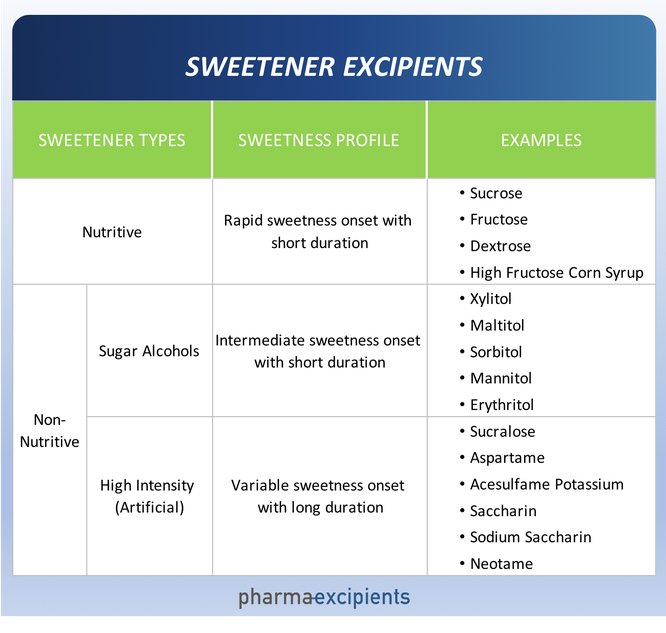
Há uma infinidade de adoçantes disponíveis para o cientista farmacêutico. Adoçantes podem ser agrupados em duas categorias - nutritivos e não nutritivos. Os adoçantes nutritivos fornecem calorias e, como o próprio nome sugere, os não-nutritivos não. Adoçantes não nutritivos podem ser ainda caracterizados como volumoso (álcoois de açúcar) e de alta intensidade (artificial).

Alguns excipientes que são doces são muitas vezes incorretamente listados em publicações como "adoçantes". Estes são excipientes usados principalmente para outros fins que não conferir doçura, por exemplo, co-solventes como glicerol ou propilenoglicol e agentes de volume tais como lactose ou maltodextrina. Os exemplos mais comuns de excipientes utilizados com a finalidade de adoçar formulações de fármacos são mostrados abaixo.
Como selecionar um adoçante
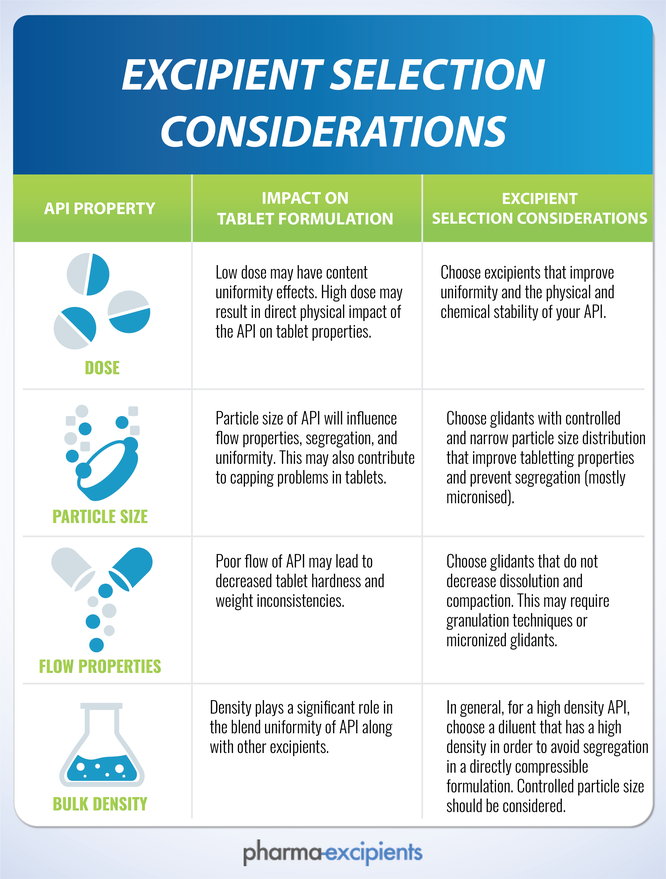
A seleção de adoçantes candidatos é informada por considerações técnicas, regulatórias e clínicas.
Considerações Técnicas
Existem inúmeros problemas técnicos que precisam ser considerados na identificação de adoçantes candidatos para o espaço de design da formulação. Esses incluem:
- Perfil de doçura. O perfil de doçura (intensidade, início e duração) de cada adoçante é diferente, conforme resumido na tabela acima. A intensidade e duração do atributo aversivo do medicamento ativo (amargo, salgado ou azedo) informa a identificação dos adoçantes candidatos. Por exemplo, muitos ativos de drogas têm um perfil de amargura de longa duração que, para palatabilidade, requer a doçura persistente proporcionada por muitos adoçantes artificiais.
- Propriedades Físicas . Adoçantes nutritivos e álcoois de açúcar têm baixa doçura relativa e às vezes são empregados como enchimentos em formas de dosagem que requerem massa adicional, como comprimidos e saquinhos de pó. Devido a esta propriedade, álcoois de açúcar e adoçantes nutritivos são conhecidos coletivamente como “adoçantes a granel”. Muitas formulações de medicamentos requerem combinações de adoçantes a granel e de alta intensidade para fornecer o perfil de doçura necessário. Alguns edulcorantes a granel têm benefícios adicionais (por exemplo, estabilidade melhorada de congelamento / descongelamento) e desvantagens (por exemplo, tendência a cristalizar em fios de tampa de garrafa de recipientes de várias doses) que também devem ser levados em consideração.
- Estabilidade Microbiológica. Dependendo do nível de uso, adoçantes a granel podem melhorar a estabilidade microbiológica, diminuindo a atividade da água, reduzindo a necessidade de conservantes químicos.
- Estabilidade química. Os adoçantes artificiais tipicamente exibem boa estabilidade no estado sólido através da placa, mas alguns têm pouca estabilidade na solução. Por exemplo, o aspartame prontamente hidrolisa em ambientes aquosos, e um produto líquido adoçado com aspartame sofreria mudanças drásticas em 6 meses de armazenamento. Alguns fabricantes não recomendam que a sucralose seja usada em formulações com pH acima de 7.
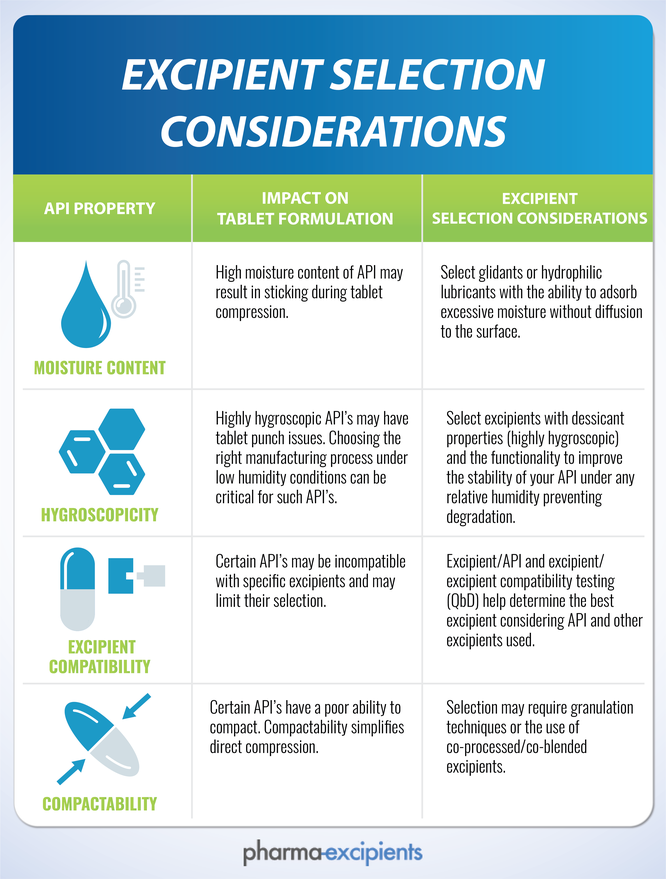
Considerações Regulamentares
A aceitação regulamentar dos adoçantes pode variar de acordo com a geografia. Muitos dos adoçantes comuns estão listados nas várias farmacopeias - Farmacopéia e Formulário Nacional dos Estados Unidos (USP-NF), Farmacopéia Britânica (BP), Farmacopeia do Japão (JP), Farmacopéia Européia (PhEur). Alguns foram determinados como sendo Geralmente Reconhecidos como Seguros (GRAS) pelo fabricante, ou pela Associação de Fabricantes de Aromatizantes e Extractos (FEMA), de acordo com a Emenda aos Aditivos Alimentares de 1958 da Lei Federal de Alimentos, Medicamentos e Cosméticos. Além disso, o FDA mantém o banco de dados de ingredientes inativos (IID), que lista os excipientes com precedentes anteriores de uso em formulações farmacêuticas aprovadas.
Um resumo de alto nível do status regulatório dos adoçantes comuns é mostrado abaixo.
Um resumo de alto nível do status regulatório dos adoçantes comuns é mostrado abaixo.

As pessoas que têm fenilcetonúria (PKU), um distúrbio genético raro, têm dificuldade em metabolizar a fenilalanina, um componente do aspartame e do advantame - um novo adoçante artificial que está quimicamente relacionado ao aspartame.
Interstingly, os produtos contendo apenas aspartame devem conter uma declaração informativa para as pessoas com PKU alertando-os sobre a presença de fenilalanina. Como o advantame é muito mais doce que o aspartame, apenas uma quantidade muito pequena precisa ser usada para atingir o mesmo nível de doçura. Como resultado, produtos contendo advantame não precisam ter essa afirmação. O Advantame é FEMA-GRAS, mas não está no IID, pois ainda não foi usado em um medicamento aprovado pela FDA, nem entrou na farmacopéia principal.
Interstingly, os produtos contendo apenas aspartame devem conter uma declaração informativa para as pessoas com PKU alertando-os sobre a presença de fenilalanina. Como o advantame é muito mais doce que o aspartame, apenas uma quantidade muito pequena precisa ser usada para atingir o mesmo nível de doçura. Como resultado, produtos contendo advantame não precisam ter essa afirmação. O Advantame é FEMA-GRAS, mas não está no IID, pois ainda não foi usado em um medicamento aprovado pela FDA, nem entrou na farmacopéia principal.
Considerações Clínicas
Considerações clínicas podem pesar contra o uso de adoçantes nutritivos devido a problemas de co-morbidade (por exemplo, diabetes) e seu potencial cariogênico, embora este último seja freqüentemente exagerado, particularmente para salvar vidas / estender medicamentos. Os álcoois de açúcar podem aumentar a motilidade gastrointestinal, mas em níveis de uso típicos, isso geralmente é apenas uma preocupação em pacientes muito jovens ou em populações especiais, como aqueles submetidos à Nutrição Parenteral Total (NPT).
A importância dos adoçantes
Para a maioria dos fármacos, o sistema adoçante desempenha um papel crucial, mas geralmente insuficiente, no desenvolvimento de formulações saborosas. Outras classes de excipientes, tais como tampões, sais, modificadores de sabor e sabores são geralmente necessários para um mascaramento eficaz do sabor.
Muitos excipientes são doces; mas apenas um subconjunto tem a função principal de transmitir doçura à formulação, ou seja, são verdadeiros adoçantes. A seleção de adoçantes é informada por uma miríade de considerações técnicas, regulatórias e clínicas. De uma perspectiva de mascaramento de sabor, o perfil de doçura - intensidade, início e duração - é extremamente importante.
Muitos excipientes são doces; mas apenas um subconjunto tem a função principal de transmitir doçura à formulação, ou seja, são verdadeiros adoçantes. A seleção de adoçantes é informada por uma miríade de considerações técnicas, regulatórias e clínicas. De uma perspectiva de mascaramento de sabor, o perfil de doçura - intensidade, início e duração - é extremamente importante.
Post do blog para pharmaexcipients.com - Preparado por Senopsys LLC por David Tisi - Todos os direitos reservados
David Tisi é o diretor técnico da Senopsys LLC , uma empresa de serviços especializados dedicada ao desenvolvimento de produtos farmacêuticos saborosos. Ele tem 15 anos de experiência em avaliação de sabor e mascaramento de sabor de drogas experimentais e aprovadas para crianças e adultos, alavancando sua experiência em ciência sensorial e química alimentar. Ele pode ser encontrado em david.tisi@senopsys.com