Seu injetável estéril está pronto para alterações nas matérias-primas?
Por Lisa Cherry, Ph.D.

Se você trabalha com um CDMO ou fabrica seu medicamento em um de seus próprios locais, provavelmente haverá alterações nas matérias-primas de tempos em tempos. Isso inclui modificações na síntese da API, principais materiais de iniciação e intermediários. Isso é esperado durante todo o ciclo de vida de um medicamento. Uma abordagem de "melhores práticas" para aceitar material alternativo de um fornecedor é vital para manter a integridade do medicamento.
Alterações nas matérias-primas podem ocorrer por vários motivos, como a adoção de avanços nos processos de fabricação, a disponibilidade de matérias-primas "equivalentes" e menos caras, ou uma alteração no local de fabricação de um produtor de matérias-primas. O pessoal de controle de matéria-prima astuto gerencia essas mudanças com cuidado. Eles sabem que uma mudança aparentemente pequena pode ter um impacto significativo no comportamento da matéria-prima em um ambiente de produção de drogas.
Os estudos de caso a seguir ilustram um processo robusto de revisão de materiais e um sistema de controle de materiais para garantir a capacidade de fabricação, qualidade e usabilidade do medicamento, mesmo quando o material alternativo se comporta de maneira um pouco diferente do original. Embora esses estudos de caso pertençam a injetáveis estéreis, os princípios descritos e a solução de problemas necessários se aplicam aos medicamentos de moléculas pequenas em geral.
Matérias-primas equivalentes
Sempre que houver uma mudança de material pendente, é útil aprender o máximo possível sobre a substância alternativa. Por exemplo, o material permanecerá na forma de flocos ou mudará para grânulos? Se for uma API, alguma das etapas do processo foi alterada? Os solventes residuais são iguais?
A comunicação com os fornecedores durante todo o processo de troca e a avaliação de quais dimensões estão mudando permite uma transição perfeita para um novo material aceitável para uso na fabricação de formas de dosagem.
Estudo de caso um: situação ideal
No Estudo de Caso Um, esboçamos um exemplo do que um gerente de controle de materiais gostaria de ver durante a qualificação de uma matéria-prima alternativa. O fornecedor da API notificou o gerente de materiais com bastante antecedência da alteração da API e forneceu amostras com tempo de entrega suficiente para avaliar o material e preparar lotes de laboratório de medicamentos para fins de comparação.
Os lotes de laboratório foram feitos com a fonte de matéria-prima atualmente aprovada e a fonte alternativa. A equipe de materiais queria ver que ambos se comportavam da mesma forma e que o processo de fabricação da forma de dosagem não seria afetado. Analiticamente, não foram encontradas diferenças detectáveis entre as APIs ou a forma de dose final. Ambas as APIs foram processadas da mesma forma no laboratório e posteriormente em grande escala. Estudos de estabilidade também confirmaram que a alteração da API não afetou a estabilidade do medicamento. O resultado final: as duas APIs se mostraram comparáveis.
Estudo de caso dois: precipitado
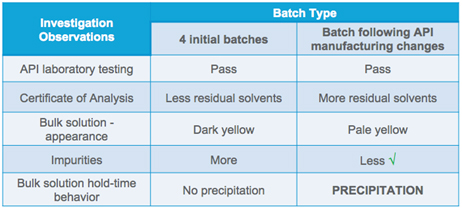
No Estudo de Caso Dois, examinamos o que pode acontecer quando uma mudança aparentemente rotineira da API apresenta um desafio no estágio de composição da produção da forma de dosagem. Como processo de rotina, a equipe de gerenciamento de materiais testou a API de entrada para identificação e comparou os resultados com o certificado de análise (COA). A equipe avançou e fabricou um lote e não observou problemas ao longo do processo.
A equipe de gerenciamento de materiais coletou amostras durante o processo de composição para realizar estudos de laboratório. E foi aí que o problema foi descoberto. Um dia após a aquisição da amostra, foi observado algum precipitado e, quanto mais as amostras permaneciam, mais precipitação se formava.
Uma investigação foi iniciada. No COA, sabia-se que níveis mais baixos de solventes residuais estavam presentes no material original e níveis relativamente mais altos no material mais novo. Durante o lançamento da API original e da nova API, no entanto, nenhuma sinalização vermelha foi levantada - ambas atenderam às especificações.
Além disso, uma comparação da solução a granel indicou níveis mais altos de impurezas com o material original, quando comparado com o novo material. Com base nesses resultados, parecia razoável que a solução em massa permanecesse estável. O comportamento do tempo de espera da solução em massa do novo material, no entanto, não correspondeu às expectativas de estabilidade.
Nesse ponto, uma discussão com o fornecedor da API foi iniciada para verificar se era possível voltar ao processo antigo. Não era. O produtor da API já havia implementado as alterações do processo da API em larga escala. As equipes de material e produção foram desafiadas a fazer a nova API funcionar.
Após uma análise abrangente, a equipe descobriu que a nova API só se solubilizaria completamente com um valor de pH específico. Portanto, foi necessário um ajuste de pH para mover a API para a solução. O ajustador de pH, no entanto, adicionou um contra-íon à API, produzindo uma forma de sal, que fez com que a substância se precipitasse da solução.
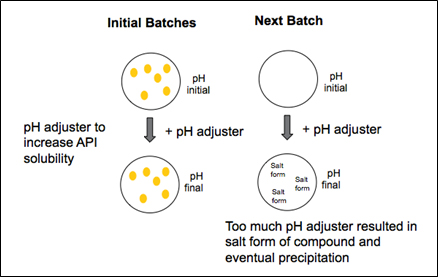
A resposta, segundo a equipe, foi reduzir a quantidade de ajustador de pH. Em vez de adicionar uma quantidade significativa no início para dissolver os excipientes, o calor foi usado para ajudar na dissolução. Os excipientes totalmente dissolvidos com o uso de calor e não foram observados efeitos adversos nas amostras mantidas quanto à estabilidade. Após o resfriamento, o pH foi ajustado usando quantidades limitadas de ajustador de pH, conforme determinado por iterações sucessivas de lotes de laboratório. Após o ajuste do pH, a API foi adicionada e misturada à solução.
Obtendo controle sobre a quantidade de ajustador de pH adicionado para produzir a solução a granel, o gerente de materiais e as equipes de produção resolveram completamente o problema do precipitado. Após a solução desse problema, o fabricante instituiu novos protocolos para aceitar qualquer material alternativo. Hoje, é necessária uma avaliação de equivalência, juntamente com estudos individuais. As avaliações de equivalência e os estudos associados, por padrão, fomentam uma discussão aberta sobre mudanças no processo que podem afetar cada parte e, ao mesmo tempo, preparam o terreno para implementar adequadamente as mudanças necessárias para garantir uma transição bem-sucedida para o novo material.
Resumo: Como se preparar para alterações de matéria-prima
Para ajudar a impedir que alterações de matéria-prima minem suas operações de fabricação, siga dois princípios básicos:
1. Trabalhe em parceria com seus fornecedores de matérias-primas. Informe-os antecipadamente que você precisa saber se eles fazem alterações em seus produtos, bem como entender completamente as alterações feitas. E;
2. Faça sua devida diligência antes de aceitar qualquer novo material. Realize testes de equivalência usando substâncias existentes e pendentes. Compare-os lado a lado, desde estudos de caracterização até produção em lotes e a granel de formas farmacêuticas acabadas.
Seguir este modelo de boas práticas servirá para manter a integridade do seu medicamento quando as matérias-primas mudarem.
Nenhum comentário:
Postar um comentário