Por Omar A. Salman, Ph.D., Conselheiro Sênior de Pesquisa, Pfizer / Pfizer CentreOne
A fabricação de suspensões aquosas estéreis requer uma compreensão completa dos fatores que influenciam sua estabilidade física e química. A morfologia das partículas do ingrediente farmacêutico ativo (API) desempenha um fator-chave na taxa de dissolução do medicamento, ressuspensibilidade e seringabilidade. O tipo e a concentração de surfactantes utilizados na formulação afetam a ressuspensibilidade do medicamento e a estabilidade química. Além disso, as tecnologias usadas na redução do tamanho de partícula API e na mistura de alto cisalhamento para a formulação de medicamentos devem ser avaliadas devido ao seu impacto nos atributos de qualidade API e medicamentos.
Uma revisão concisa desses fatores é apresentada neste artigo.
Processo de manufatura
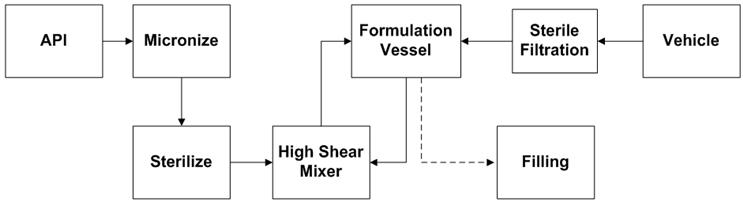
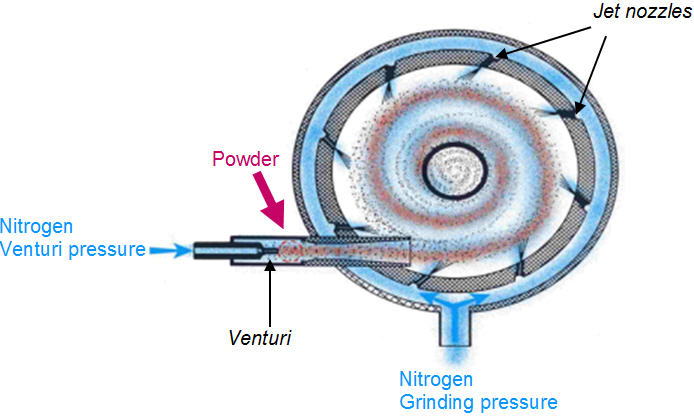
Um diagrama de fluxo das etapas principais na fabricação de suspensões aquosas estéreis é ilustrado na Figura 1. O tamanho de partícula da API cristalizada assepticamente (não moída) é reduzida usando um moinho de jato de fluido, ou micronizador, para a distribuição de tamanho de partícula desejada perfil. Em um moinho de jato fluido (Figura 2), a redução de tamanho é alcançada por colisão de partícula para partícula. As partículas de pó são alimentadas na câmara de moagem por um sistema Venturi. O gás de jato (nitrogênio ou ar) entra através de um conjunto de bicos De-Laval que aumentam a aceleração das partículas para atingir a velocidade supersônica (300-500 m / s). A colisão entre as partículas que entram na câmara de moagem e as partículas que se movem em espiral dentro da câmara resulta na quebra de partículas. Devido à força centrífuga, partículas mais finas saem pela parte central da câmara, enquanto partículas maiores na área do anel externo continuam a acelerar até que seu tamanho seja reduzido ainda mais por colisão [1]. Os principais parâmetros que afetam o tamanho das partículas são a taxa de alimentação do pó e a pressão do fluido do jato (pressão de moagem). Aumentar a taxa de alimentação aumenta a concentração do produto na câmara do micronizador, reduzindo assim o espaço de aceleração entre as partículas. Em geral, taxas de alimentação mais altas resultam em partículas mais grossas. Maior pressão de moagem significa maior energia de micronização, que produz partículas mais finas. Aumentar a taxa de alimentação aumenta a concentração do produto na câmara do micronizador, reduzindo assim o espaço de aceleração entre as partículas. Em geral, taxas de alimentação mais altas resultam em partículas mais grossas. Maior pressão de moagem significa maior energia de micronização, que produz partículas mais finas. Aumentar a taxa de alimentação aumenta a concentração do produto na câmara do micronizador, reduzindo assim o espaço de aceleração entre as partículas. Em geral, taxas de alimentação mais altas resultam em partículas mais grossas. Maior pressão de moagem significa maior energia de micronização, que produz partículas mais finas.
A API micronizada é embalada em bolsas Tyvek ® e depois esterilizada usando um esterilizante a gás, por exemplo, óxido de etileno. A esterilização por irradiação gama ou feixe de elétrons pode ser usada apenas se essas tecnologias não tiverem impacto na qualidade do produto. Por exemplo, geralmente, a irradiação não é viável para corticosteróides devido à degradação radiolítica [2]. A API esterilizada terminalmente é então adicionada assepticamente em uma área de Grau A ao veículo estéril, e a suspensão é misturada usando um misturador de alto cisalhamento para dispersar e molhar completamente as partículas.
A etapa final do processo de fabricação é o preenchimento. Os frascos pré-esterilizados são preenchidos com um volume fixo de suspensão, usando um sistema totalmente automatizado, e selados com uma tampa e uma tampa.
Figura 1: Fluxograma do processo de formulação de suspensão estéril
Figura 2: Diagrama esquemático do processo de micronização usando um moinho de jato de fluido [1]
Características da API
O tamanho das partículas desempenha um papel fundamental na estabilidade das suspensões. As partículas grandes têm uma taxa de sedimentação mais rápida, de acordo com a lei de Stokes, e podem entupir a agulha durante a retirada da suspensão do frasco ou após a injeção, resultando em dosagem inadequada. As partículas finas, por outro lado, formam um bolo no fundo do frasco que é difícil de dispersar se não forem adequadamente colocadas em ponte durante a formulação.
Além disso, a distribuição do tamanho das partículas afeta a taxa de dissolução da substância medicamentosa, conforme descrito pela equação de Noyes-Whitney:
dC / dt = (C s- C) DA / Vh (1)
onde, dC / dt = taxa de dissolução, D = coeficiente de difusão, A = área superficial da partícula, C s = solubilidade, C = concentração no tempo t, V = volume da solução e h = espessura da camada limite. À medida que o tamanho das partículas é reduzido, a área superficial aumenta, aumentando a taxa de dissolução e, consequentemente, a biodisponibilidade.
Formulação do veículo
Os principais componentes de um veículo de base aquosa são:
- um agente umectante para substituir a interfase ar-sólido por uma interfase líquido-sólido
- um surfactante para formar uma suspensão termodinamicamente estável, superando as forças atrativas do tipo Van Der Waals entre partículas
- cloreto de sódio para isotonicidade
- um conservante como álcool benzílico
- antioxidante e
- um buffer se for necessário controle de pH
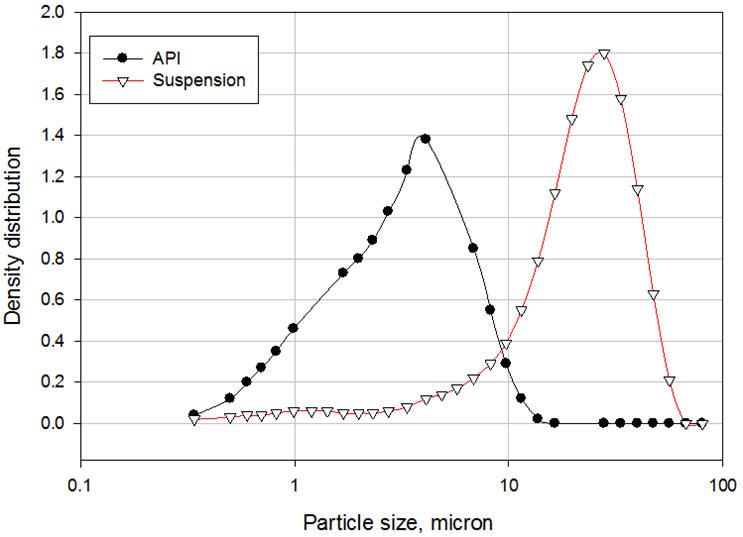
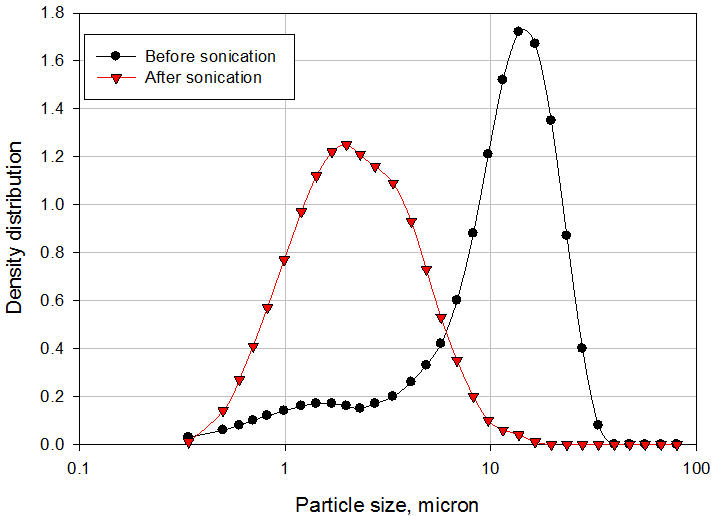
O polietilenoglicol 3350 (PEG) é um surfactante solúvel em água não-iônico que possui a fórmula química de HO (CH 2 CH 2 O) nH e é comumente usado na formulação de veículo para fornecer estabilização estérica da suspensão. Segmentos de polímero PEG, chamados de cadeias de "ancoragem", absorvem a superfície das partículas de API para formar uma camada de adsorção. A espessura desta camada depende de vários parâmetros, como concentração de polímero, solvência do meio, temperatura e peso molecular do polímero. Os outros segmentos, chamados de cadeias estabilizadoras ou "caudas", se estendem para a solução [3]. Essas caudas se interconectam para formar uma ponte entre as partículas, resultando em floculação controlada. Uma comparação entre a distribuição do tamanho de partícula da API antes e depois da formulação ilustra claramente o fenômeno da ponte (Figura 3). Observe que as suspensões que contêm partículas extremamente finas (nanopartículas) são geralmente estáveis e não requerem a adição de surfactantes. O movimento browniano das partículas neutraliza a força gravitacional, de modo que as partículas permaneçam suspensas na mídia. À medida que as partículas se tornam mais grossas, sedimentam e formam sedimentos compactos, difíceis de dispersar novamente se nenhum surfactante for adicionado ao veículo [4]. Se um surfactante for adicionado, no entanto, o pó se deposita como partículas com pontes frouxas que são fáceis de dispersar novamente.
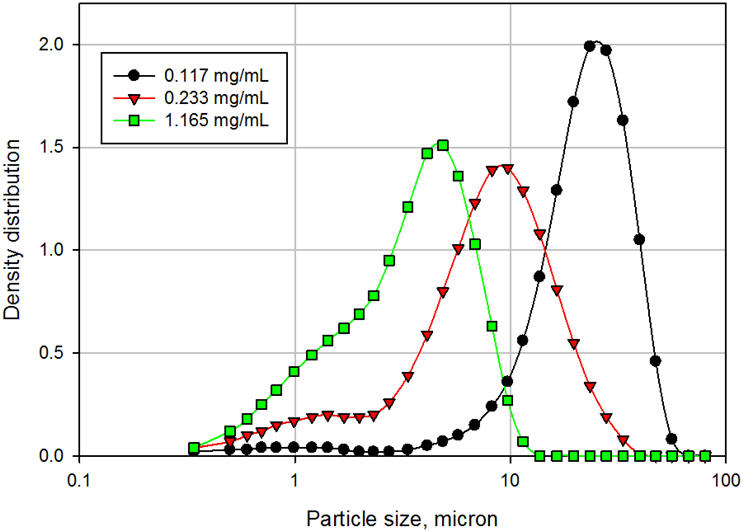
A concentração de surfactante desempenha um papel crítico na redispersibilidade da substância farmacológica. Adicionar mais do que a quantidade ideal pode ter efeitos adversos, como o endurecimento. A Figura 4 compara a distribuição do tamanho de partícula das suspensões em níveis crescentes de um surfactante iônico. O tamanho médio de partícula da suspensão diminuiu de 23,5 µ para 9,2 µ e para 3,9 µ, à medida que a concentração de surfactante aumentou de 0,117 mg / mL, para 0,233 mg / mL e 1,165 mg / mL, respectivamente. Uma tendência semelhante foi observada para a altura estabelecida do fármaco (SDH), definida como o volume da API estabelecida sobre o volume total da suspensão e é considerado um marcador de redispersibilidade. O SDH diminuiu de 51% na concentração de surfactante de 0,117 mg / mL para 12% na concentração de surfactante de 1,165 mg / mL. Mais distante,
Figura 3: Distribuição do tamanho de partícula da API e suspensão
Figura 4: Distribuição granulométrica das suspensões em função da concentração de surfactante
Sabe-se que o PEG e surfactantes semelhantes, como os polissorbatos, são suscetíveis à auto-oxidação para formar hidroperóxidos, seguidos pela degradação da cadeia em subprodutos, como o ácido fórmico. Donbrow et al. [5] mostraram que as soluções aquosas de Polissorbato 20 se degradam devido à autoxidação e a degradação está associada a um aumento no número de peróxidos e uma queda no pH devido à formação de ácidos. A taxa de formação de ácido aumentou a temperaturas mais altas. Em estudo semelhante, Donbrow et al. [6] concluíram que os ácidos fórmico e acético foram formados devido à degradação da extremidade hidrofílica do polioxietileno dos surfactantes não iônicos. Estes ácidos são formados na etapa de terminação da degradação de radicais livres da porção oxietileno. Concluiu-se também que a taxa de formação de ácido aumentou a uma temperatura de incubação mais alta.
O seguinte mecanismo foi sugerido:
| Iniciação |
RH → R . + H .
|
| Propagação |
R . + O 2 → ROO .
ROO . + RH → ROOH + R .
|
| Terminação | 2ROO . →
ROO de produtos inativos . + R. → Produtos inativos |
Na etapa de iniciação, os radicais livres são formados devido à luz, calor, iniciadores químicos ou catalisadores. No segundo passo, a propagação, a base de carbono radical (R . ) Reage com o oxigénio, formando um peróxido orgânico (ROO . ), O qual reage com o substrato (RH) para produzir um ácido e um novo radical de carbono de repetir os passos de propagação . Na etapa de terminação, os radicais livres são desativados por colisões bimoleculares [5-7].
Além da queda de pH, a degradação do surfactante pode resultar em espessamento da suspensão, levando a problemas de uniformidade do conteúdo. Para resolver a queda de pH causada pela degradação oxidativa do PEG, o ar no espaço superior do frasco é substituído por nitrogênio, ou um agente tampão é adicionado à formulação.
Mistura de alto cisalhamento
Para molhar e dispersar completamente a API no veículo, é necessária uma mistura de alto cisalhamento para que os surfactantes possam efetivamente adsorver na superfície de cada partícula. Vários tipos de misturadores de alto cisalhamento podem ser usados. Os misturadores do tipo rotor-estator de lote (por exemplo, IKA ® , Silverson ® , Ystral ® ) são geralmente utilizados em laboratórios, pilotos e embarcações de produção de pequeno volume. Para equipamentos de grande escala, misturadores do tipo em linha, como Tri-Blender ® ou Ystral TDS ®(sistema de transporte e distribuição). No projeto Tri-Blender [8], uma bomba centrífuga é usada para puxar o pó de uma tremonha localizada na parte superior através de um tubo difusor para uma câmara de mistura onde o impulsor está localizado. O veículo é bombeado do tanque de formulação e entra tangencialmente através de um tubo externo para a câmara de mistura. A suspensão é então devolvida ao tanque. No sistema TDS [9], o pó é introduzido verticalmente por alto vácuo gerado por um rotor de alta velocidade. O líquido (veículo) é retirado do fundo do tanque de formulação para a câmara de distribuição do lado oposto da entrada de pó. Na câmara de dispersão, o pó é disperso no líquido sob taxas de cisalhamento muito altas e a suspensão é reciclada no tanque.
Uma nova tecnologia promissora que pode ser usada para mistura de alto cisalhamento e redução do tamanho de partícula é o Microfluidizer ®Processador. Uma vantagem importante dessa tecnologia é a eliminação da etapa de micronização que exige muito trabalho. Por conseguinte, a redução e a formulação do tamanho podem ser realizadas em uma etapa. O componente central desta tecnologia é a câmara de interação. A câmara possui microcanais com dimensões tão pequenas quanto 50µ através das quais o fluido flui a velocidades de até 500 m / s. O exterior da câmara é feito de aço inoxidável, enquanto o interior é feito de diamante ou cerâmica. Um diagrama esquemático do processador de microfluidizador é mostrado na Figura 5 [10]. O veículo e as partículas grosseiras de API são adicionadas a um recipiente de alimentação. A bomba intensificadora empurra a suspensão através da câmara de interação a pressões de até 40.000 psi. As altas forças de cisalhamento, a colisão partícula-partícula e a colisão na parede partícula resultam na redução do tamanho da partícula. A suspensão é resfriada por um trocador de calor e é coletada em um recipiente receptor ou reciclada no recipiente de alimentação para mais passagens, dependendo do tamanho da partícula alvo. Esta tecnologia pode reduzir o tamanho das partículas para menos de 1 µ.
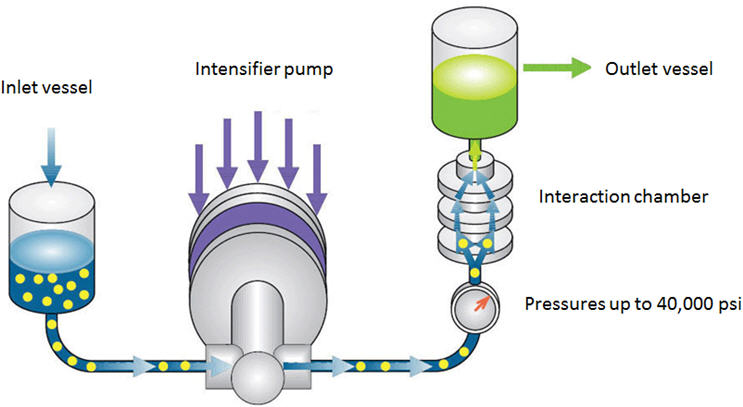
Figura 5: Diagrama esquemático do processador do microfluidizador [10]
Um exemplo típico da aplicação do processador de microfluidizador na fabricação de suspensões é discutido aqui. O veículo e o pó API não moído grosso foram adicionados a um recipiente de alimentação agitado do Processador de Microfluidizador em escala piloto modelo 110-EH. A suspensão foi recirculada através da câmara de interação H10Z, que possui micro canais de 100μ a uma pressão de 20.000 psi por 6 minutos. As amostras foram removidas após 2, 4 e 6 minutos de recirculação para medir a distribuição do tamanho de partícula por difração a laser. Para medir o tamanho das partículas primárias, as amostras foram sonicadas por 30 segundos para quebrar a ponte entre as partículas. Os resultados são mostrados na Figura 6. O tamanho médio de partícula diminuiu de 157,71µ para a API não fresada para 5,89µ após apenas 2 minutos de recirculação. Após 4 e 6 minutos de recirculação, o tamanho médio das partículas caiu para 3,35µ e 2,73µ, respectivamente. A suspensão final teve excelente redispersibilidade, exigindo apenas 1 inversão para suspender completamente a API após 24 horas de assentamento. Além disso, a suspensão passou no teste padrão de seringabilidade. A ponte das partículas na suspensão final é claramente ilustrada na Figura 7. O tamanho médio de partícula da suspensão antes da sonicação (como está) foi de 12,31 µ. Após sonicação, o tamanho médio das partículas era de 2,73 µ, o que representa o tamanho das partículas primárias. A ponte das partículas na suspensão final é claramente ilustrada na Figura 7. O tamanho médio de partícula da suspensão antes da sonicação (como está) foi de 12,31 µ. Após sonicação, o tamanho médio das partículas era de 2,73 µ, o que representa o tamanho das partículas primárias. A ponte das partículas na suspensão final é claramente ilustrada na Figura 7. O tamanho médio de partícula da suspensão antes da sonicação (como está) foi de 12,31 µ. Após sonicação, o tamanho médio das partículas era de 2,73 µ, o que representa o tamanho das partículas primárias.
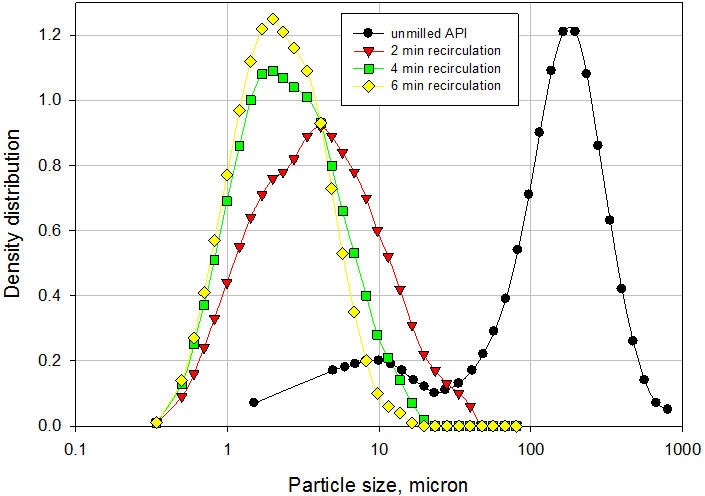
Figura 6: Distribuição do tamanho de partícula da suspensão formulada usando o processador de microfluidizador
Figura 7: Distribuição do tamanho de partícula de partículas primárias e em ponte
Simulação de dinâmica molecular
A simulação da dinâmica molecular (MD) é um método em mecânica estatística que envolve a solução da segunda lei do movimento de Newton sob certas restrições para todos os átomos e moléculas no sistema em estudo. As simulações de MD podem ser usadas para prever o mecanismo de adsorção e a cinética de um surfactante na superfície dos cristais de API. Utilizando o campo de força CHARMM36 [11] e o servidor CgenFF [12] para os parâmetros e arquivos de topologia do PEG e da API, foram realizadas simulações de MD para investigar a adsorção de cadeias de 25-PEG na superfície de um cristal de corticosteróide. Instantâneos de moléculas de PEG com todos os seus átomos em vermelho em uma solução aquosa no tempo = 0 e após 6 nanossegundos são ilustrados na Figura 8 (em cima) e (em baixo), respectivamente. Contas cinzentas representam átomos nas moléculas de corticosteróides em placas de cristal de ambos os lados. As moléculas de água não são mostradas para maior clareza. Essas simulações suportam claramente o mecanismo discutido anteriormente. Um lado da molécula de PEG (provavelmente o hidrofóbico) adsorve na superfície do cristal, enquanto o outro lado (provavelmente o hidrofílico) se estende no líquido.
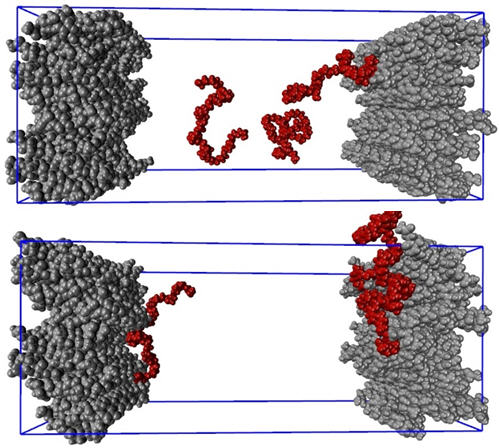
Figura 8: Adsorção da cadeia PEG de 25-mer (vermelho) no cristal API (pilhas cinza de ambos os lados)
Sumário
As principais operações da unidade na fabricação de suspensões aquosas estéreis são a redução do tamanho das partículas, a formulação do veículo, a esterilização e a mistura de alto cisalhamento. Todas essas operações podem afetar as propriedades, bem como a estabilidade das suspensões. A taxa de dissolução e a taxa de sedimentação da substância do medicamento são uma função do tamanho das partículas. O tipo e a concentração de surfactantes utilizados na formulação influenciam a ressuspensibilidade da suspensão. A mistura de alto cisalhamento é essencial para umedecer e dispersar as partículas durante a formulação.
Referências
- www.dec-group.net
- MP Kane, K. Tsuji. Esquema de degradação radiolítica de 60 corticosteróides co-irradiados. Journal of Pharmaceutical Sciences, 72 (1), 1983.
- T. Tadros. Forças de interação entre partículas contendo camadas de polímero enxertado ou adsorvido. Avanços na Ciência da Interface coloidal, 104, 2003.
- T. Tadros. Controle de estabilidade / floculação e reologia de suspensões concentradas. Pure & Appl. Chem., 64 (11), 1992.
- M. Danbrow, E. Azaz, A. Pillersdrof. Autoxidação de polissorbatos. J. Pharm. Sei. 67 (12), 1978.
- M. Danbrow, R. Hamburger, E. Azaz, Pillersdorf. Desenvolvimento de acidez em surfactantes não iônicos: ácido fórmico e acético. Analyst, 103, 1978.
- B. Kerwin. Polissorbato 20 e 80 Usado na Formulação de Bioterapêuticos de Proteínas: Estruturas e Vias de Degradação. J. Pharm. Sci, 97 (8), 2008.
- Boletim técnico. Tri-Clover, Inc.
- http://ystral.com/
- www.microfluidicscorp.com .
- http://mackerell.umaryland.edu/charmm_ff.shtml#gromacs
- https://cgenff.paramchem.org/