À medida que a indústria biofarmacêutica continua avançando seus métodos de tratamento de doenças, novos sistemas de administração de medicamentos estão sendo desenvolvidos para atingir áreas específicas de um paciente, em vez de administrar o tratamento sistemicamente. Por exemplo, no caso de conjugados de drogas com anticorpos (ADCs), o medicamento para câncer citotóxico é conjugado com o anticorpo para que possa ser recebido apenas pelo tumor e não pelo resto do corpo. Isso reduz os efeitos colaterais prejudiciais ao paciente e aumenta a eficácia do medicamento. As formulações para esses tipos de sistemas de administração de medicamentos são complexas e, portanto, apresentam desafios em relação à estabilidade, eficácia, potência e até segurança dos medicamentos durante as fases clínica e comercial. Freqüentemente, a implementação de equipamentos e / ou processos especializados é necessária para garantir uma formulação bem-sucedida.
Este artigo discute seis tipos principais de formulações complexas, bem como os equipamentos e processos importantes necessários para desenvolver processos compatíveis com GMP. Qualquer empresa que adote um sistema de administração de medicamentos que envolva uma formulação complexa deve estar preparada para os requisitos relacionados a esse tipo de desenvolvimento de medicamentos, a fim de obter expansão e fabricação bem-sucedidas.
FORMULAÇÕES COMPLEXAS COMUNS IMPLEMENTADAS HOJE
Uma formulação é considerada complexa se o processo exigir etapas adicionais além da simples mistura e filtração. Abaixo estão algumas das formulações complexas mais amplamente usadas na indústria farmacêutica atualmente. Cada formulação é única nos desafios de fabricação que apresenta, mas algumas soluções podem ser compartilhadas.
1. SUSPENSÕES
Uma suspensão é uma mistura heterogênea que contém partículas sólidas suficientemente grandes para sedimentação. Existem três tipos principais de suspensões:
► EMULSÕES DE ÓLEO / ÁGUA
Uma emulsão é uma mistura de dois ou mais líquidos que são normalmente imiscíveis (não misturáveis ou não misturáveis). Os termos colóide e emulsão são às vezes usados de forma intercambiável. Em uma emulsão, um líquido é disperso no outro. Dois líquidos podem formar diferentes tipos de emulsões. Como exemplo, óleo e água podem formar uma emulsão de óleo em água, onde o óleo é suspenso em água. Também pode ser formada uma emulsão de água em óleo, onde a água é dispersa no óleo. Também são possíveis várias outras emulsões, incluindo uma emulsão "água em óleo em água" e uma emulsão "óleo em água em óleo".
Como as emulsões são líquidas, elas não exibem uma estrutura interna estática. Presume-se que as gotículas dispersas na matriz líquida (denominadas meio de dispersão) sejam distribuídas estatisticamente. Microemulsões são usadas para administrar vacinas. Emulsões típicas usadas nessas formulações são nanoemulsões. O óleo é emulsionado com detergentes usando um misturador de alto cisalhamento para estabilizar a emulsão; portanto, quando os óleos encontram lipídios na membrana celular ou no envelope de bactérias ou vírus, eles forçam os lipídios a se fundirem. Alguns exemplos de medicamentos formulados como emulsões de óleo / água são Diprivan® (propofol), um anestésico injetável, e Restasis® (ciclosporina), uma gota oftálmica para a síndrome do olho seco. Outras aplicações de nanoemulsões incluem o tratamento de infecções do sistema retículo-endotelial,
Existem três processos usados em emulsões de óleo / água:
Mistura de alto cisalhamento - dispersa ou transporta ingredientes monofásicos para uma fase contínua com a qual normalmente seria imiscível. Existem dois tipos de equipamento de mistura para alto cisalhamento: overhead, usado para lotes com 20 litros ou menos, ou em linha, que é usado para lotes com mais de 20 litros.
Homogeneização - mistura intensiva de substâncias ou grupos mutuamente relacionados para formar uma constante de diferentes fases insolúveis para obter uma suspensão ou emulsão. O objetivo da homogeneização é diminuir o tamanho das partículas e aumentar a estabilidade.
Extrusão - um processo usado para criar objetos de um perfil transversal fixo. Um material é empurrado ou puxado através de uma matriz da seção transversal desejada. Para produtos farmacêuticos, a extrusão através de filtros poliméricos nanoporosos é usada para produzir suspensões de lipossomos ou transferossomos de um tamanho específico e uma distribuição estreita de tamanho. Por exemplo, o medicamento anti-câncer Doxorrubicina no sistema de entrega de lipossomas é formulado por extrusão. A reprodutibilidade, a capacidade de obter um tamanho de partícula uniforme até as dimensões submicrônicas, é uma das principais vantagens. Isso resulta com mais freqüência na obtenção da qualidade desejada do produto em uma única passagem.
A mistura e a homogeneização de alto cisalhamento geralmente são realizadas em salas limpas IS0 7 e são consideradas operações não estéreis. Se uma operação de formulação estéril for necessária, ela deverá passar pela validação asséptica do processo. O padrão da indústria para uma membrana filtrante de esterilização é de 0,22 micrômetros ou 220 nanômetros (nm). Partículas suspensas maiores que 220 nm não podem ser filtradas estéreis e, portanto, requerem técnicas de formulação asséptica. Qualquer coisa estéril também deve ser validada e testada.
►LIPOSSOMOS
Um lipossoma é uma vesícula preparada artificialmente composta por uma bicamada lipídica. Eles podem ser usados como veículo para administração de nutrientes e medicamentos. Os lipossomas são difíceis de fabricar e requerem solventes para dissolver o lipídio. A razão pela qual as drogas são encapsuladas em lipídios é o princípio básico, "como dissolve-se". Os pulmões, por exemplo, possuem lipídios em torno dos bronquíolos, o que lhes permite expandir e contrair durante a inspiração / expiração. Os sistemas de entrega lipossômica oferecem vantagens importantes porque são mais facilmente absorvidos em uma célula e permitem a liberação sustentada de API a partir de sistemas de entrega lipossômica. É possível que alguns tratamentos contra o câncer sejam distribuídos usando esse método como uma maneira de fornecer medicamentos diretamente para as áreas onde as células cancerígenas estão localizadas. Você pode aquecê-las in situ e "quebrar" o lipídio para liberar a droga. Também é usado para uma maior absorção de drogas tóxicas nos locais-alvo, reduzindo assim o risco de uma exposição sistêmica a drogas tóxicas apenas para observar seus efeitos nos pacientes.
Os seguintes processos podem ser usados na produção de lipossomas:
Encapsulamento - Existem três maneiras de encapsular um lipossoma. Eles são:
- a infusão de dois ou mais fluxos de solução
- sonicação, que é o ato de aplicar energia sonora para agitar partículas em uma amostra para diversos fins
- mistura de alto cisalhamento.
Filtragem de fluxo tangencial (TFF) - TFF é um tipo de filtragem na qual a alimentação é passada através de uma membrana ou leito. Os sólidos são retidos pelo filtro e o filtrado é liberado "tangencialmente" do volume recirculado. No TFF, o fluido é bombeado através da superfície da membrana. Uma pressão transmembranar aplicada serve para forçar uma porção do fluido através da membrana para o lado do filtrado. Isso permite TFF de uso único, o que elimina a necessidade de validação de limpeza e oferece a capacidade de atingir uma especificação de concentração restrita.
Validação - É necessária uma validação de processo asséptico (APV) para mostrar que o processo pode ser realizado sem comprometer a integridade estéril do produto, tornando-o seguro para uso humano. Também são realizados vários ensaios de CQ para verificar se o produto é bom para uso. Por exemplo, uma simulação de processo é realizada para demonstrar que a esterilidade do volume formulado não está sendo comprometida a qualquer momento. Isso é alcançado usando meios líquidos nutritivos em vez de API e excipientes.
► nanopartículas
As nanopartículas são vagamente definida como substâncias com uma dimensão do produto de menos de 100 nm, mas a categoria, particularmente no domínio dos produtos farmacêuticos, inclui substâncias que são tão grandes como 500 nm. Para um ponto de comparação, um cabelo humano tem aproximadamente 80.000 nm de largura. Nanopartículas têm sido usadas na administração de medicamentos há muitos anos. Por exemplo, o Abraxane® utiliza a liberação de nanopartículas do agente anticâncer Paclitaxel (Taxol). Os desenvolvedores de medicamentos podem exigir que os lipídios sejam fabricados e dimensionados para nanopartículas, a fim de permitir uma melhor absorção do medicamento pelo organismo. Isso geralmente é feito por microfluidização, que é essencialmente homogeneização usando microfluídicos.
Existem várias considerações que precisam ser feitas quando se trata de nanopartículas, como:
- A formulação da nanopartícula pode exigir a mistura das fases aquosa e oleosa e depois homogeneizar com uma especificação de tamanho de partícula.
- Se um novo equipamento for introduzido para formulação, instalação, operacional e qualificação de desempenho (IQ / OQ / PQ), podem ser necessários protocolos de limpeza e esterilização. Isso garante que o equipamento esteja funcionando corretamente e que todos os parâmetros do processo estejam em um estado de controle qualificado.
- Se o tamanho médio das partículas for maior que 220 nm, é provável que seja necessário processamento asséptico.
- A natureza e a gravidade de qualquer resíduo devem ser consideradas. Se for determinado que o resíduo representa um perigo, ele deve ser coletado em contêineres aprovados pelo Departamento de Transportes (DOT) e manuseado somente por operadores qualificados. Protocolos específicos devem ser seguidos para garantir o descarte adequado.
2. CONJUGADOS DE DROGAS
Conjugados de medicamentos consistem em um medicamento ligado covalentemente a outra molécula. Alguns exemplos incluem drogas conjugadas a proteínas ou polímeros. Um dos tipos mais promissores de conjugados são os ADCs (como mencionado acima). ADCs consistem em anticorpos monoclonais ligados a uma droga biologicamente ativa por meio de um ligante químico. Como mencionado acima, ao vincular um medicamento citotóxico a um anticorpo, a quantidade de material citotóxico absorvido pelo resto do corpo é reduzida ao direcionar especificamente receptores para as células cancerígenas. Os ADCs são difíceis de produzir e são predominantemente usados em tratamentos contra o câncer.
Com ADCs, é importante avaliar a toxicidade do conjugado e dos agentes de ligação. Muitas vezes, os solventes devem ser adicionados e subtraídos da mistura para que a ligação ocorra. Devido à potência desses compostos, é necessária alta contenção para proteger o meio ambiente e os técnicos. Um exemplo do processo usado para conjugar um ligante a um anticorpo usando processamento asséptico é descrito mais adiante neste artigo.
3. PROTEÍNAS CRISTALIZADAS
Quando uma solução na qual uma proteína está sendo dissolvida se torna supersaturada, as proteínas se formam em cristais. As moléculas de proteína individuais criadas sob essas condições são agrupadas em uma matriz de repetição, que são mantidas juntas por interações não covalentes. Existem dois desafios principais no desenvolvimento de uma formulação de suspensão de cristal. O primeiro é o desafio de encontrar uma condição robusta de cristalização que produza cristais em menos de 24 horas, o que é suficientemente curto para a fabricação de GMP. O segundo desafio é o desenvolvimento de uma formulação de medicamento adequada para injeção, mantendo a estabilidade na estrutura cristalina. As proteínas cristalizadas podem abordar questões de viscosidade, potencialmente fornecer liberação sustentada e melhorar a seringabilidade e a injetabilidade que são frequentemente associadas a formulações de proteínas solúveis.
4. ANTICORPOS MONOCLONAIS (mAbs)
Os pipelines de desenvolvimento de medicamentos estão cheios de mAbs, e vários desses produtos se tornaram alguns dos medicamentos mais vendidos no mundo. Normalmente, são necessárias altas doses de mAbs para alcançar o benefício clínico desejado; no entanto, a viscosidade de soluções com alta concentração de mAbs apresenta desafios durante a fabricação e a administração.
Além disso, como com outros biofarmacêuticos à base de proteínas, a terapêutica à base de mAb é suscetível à degradação. Isso apresenta desafios de estabilidade em toda a cadeia de suprimentos, bem como durante o armazenamento a longo prazo e a administração de medicamentos. Antes da formulação, as vias de degradação e os riscos ambientais para a molécula devem ser identificados. Para reduzir a degradação das moléculas durante a formulação, o tampão, o pH e os excipientes devem ser otimizados.
FORMULAÇÕES ASSÉPTICAS VS NÃOASEÉPTICAS
O processamento asséptico é o manuseio de componentes de medicamentos, sistemas de fechamento de recipientes e excipientes, a fim de evitar a contaminação microbiana. O processo usado para uma formulação é asséptico ou não asséptico, o que depende se o produto final é ou não filtrável por esterilização. Por sua vez, isso determina que classe de sala limpa é necessária. As salas limpas de formulação geralmente contêm equipamentos, como capas de fluxo de ar laminar, isoladores, filtros de esterilização, autoclaves e túneis de despirogenação.
Formulações envolvendo o uso de solventes orgânicos são um exemplo de quando o processamento asséptico seria necessário. Solventes orgânicos são usados em certas formulações para dissolver lipídios ou outros componentes hidrofóbicos da formulação que são insolúveis em soluções aquosas. Alguns exemplos incluem etanol, dimetilsulfóxido (DMSO) e metanol. Esses solventes apresentam sérios riscos devido à sua inflamabilidade e exigem um projeto de engenheiro muito estratégico, pois a chama introduzida em um conjunto deve ser protegida contra explosão. Um sistema deve ser capaz de detectar o risco de ignição e desligar antes que esse risco resulte em perigo para a equipe e para o produto.
O exemplo abaixo ilustra a execução de um processo para um lipossoma asséptico em um espaço não asséptico, usando um sistema totalmente contido e totalmente fechado que é esterilizado e validado.
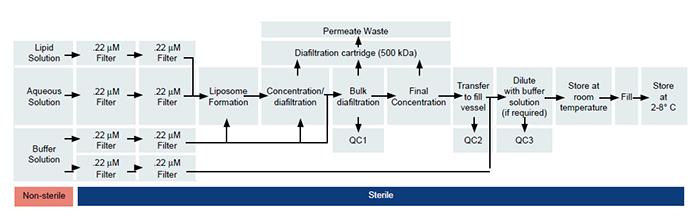
O processo começa com três soluções separadas: uma solução lipídica de etanol inflamável, uma solução antibiótica em água e uma solução salina, que é utilizada diafiltração. As três soluções são filtradas separadamente em um recipiente estéril (um tanque de aço inoxidável que foi cozido no vapor e esterilizado) usando filtração de uso único irradiada por gama. Cada um deles é filtrado separadamente no tanque. Tudo a jusante dessa solução ocorre em um processo asséptico em que um concentrado é diafiltrado. Como o lipossomo é mais do que algumas centenas de nanômetros, o produto é transferido para fora do skid usando a tecnologia de conectores assépticos de maneira estéril, para que não seja necessária mais filtragem. O processo é uma síntese de aço inoxidável para limpeza e esterilização no local, juntamente com o uso de sacos de uso único, tubulações e filtros,
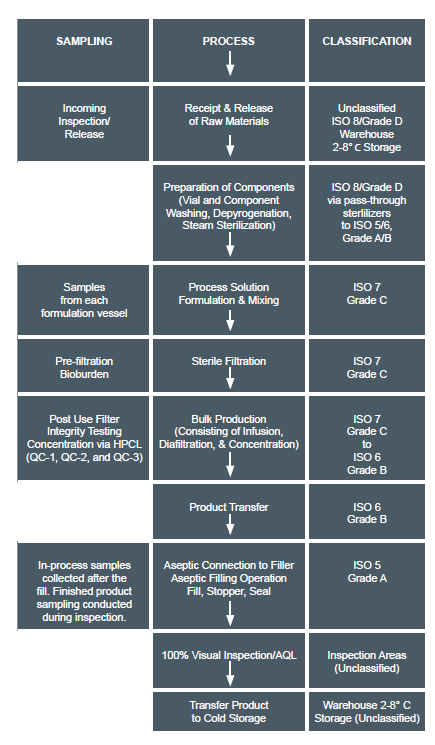
Outro exemplo de uma formulação asséptica sob condições não assépticas está acima. Isso mostra uma conjugação de anticorpos e diafiltração por ultrafiltração (UF / DF). É um TFF realizado em condições não-assépticas porque pode ser filtrado após a formulação, enquanto formulações lipossômicas que não podem ser filtradas após são concluídas em condições assépticas.
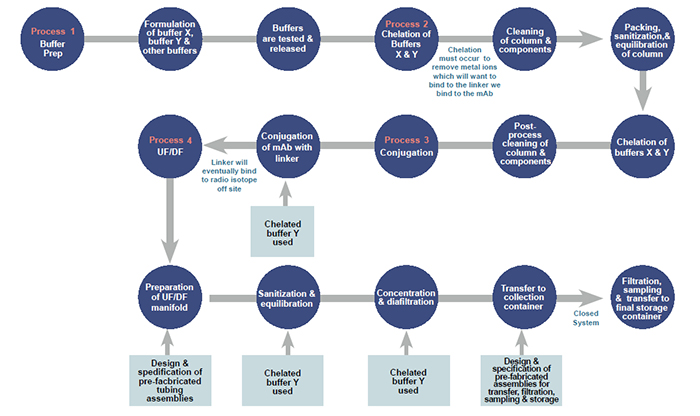
Geralmente, quando uma formulação complexa é feita em um ambiente asséptico, ela vai direto para o preenchimento assim que a formulação é concluída. No entanto, etapas adicionais devem ser concluídas para formulações feitas em ambientes não assépticos, ilustradas no exemplo na próxima página. Nesse caso, os buffers X e Y estão sendo preparados para parte de um processo maior. Existem várias etapas usando vários excipientes que foram feitos anteriormente para que os tampões possam ser formulados posteriormente.
Os buffers são quelatados para remover os íons metálicos, porque os íons metálicos competirão com o ligante que será conjugado no mAb posteriormente. Depois que os buffers são quelatados nos primeiros dias do processo, a etapa de conjugação é implementada no terceiro dia e faz uso do Tampão X quelatado. Após a conclusão da conjugação, a etapa de ultrafiltração e diafiltração (UF / DF) é a próxima , que requer outro dos buffers previamente quelatados.
Esse anticorpo conjugado é então concentrado e diafiltrado usando conjuntos de tubos pré-fabricados, um suporte de cassete TFF e membrana de filtro de cassete TFF apropriada. O tampão quelatado Y é então usado para fazer a porção de diafiltração seguida pela transferência do material através do uso de conjuntos de tubos pré-fabricados adicionais. Finalmente, essa transferência e diluição final do sistema fechado é seguida por outra transferência / filtração e, em seguida, amostragem asséptica utilizando seccionadores assépticos. A filtragem final pode ser concluída posteriormente. Enquanto isso, ele é armazenado em um recipiente de armazenamento pré-montado a uma temperatura muito específica.
RESUMO
Devido à complexidade e sensibilidade de formulações complexas, nem todas as empresas têm os recursos necessários para concluir esses processos sofisticados sob as condições apropriadas para a fabricação de cGMP. Fazer isso com sucesso requer um investimento considerável em equipamentos e mão de obra qualificada. Uma empresa também deve estar pronta para apresentar seus processos aos reguladores para ensaios clínicos e arquivamento comercial.
CDMOs experientes que oferecem uma formulação de balcão único, além de fabricação de acabamento de acabamento, oferecem transferências completamente contínuas de uma fase para a próxima, tudo em um único local. Ao fazer isso, os riscos da cadeia de suprimentos e os erros de comunicação e processo associados às transferências entre vários fornecedores podem ser minimizados ou mesmo eliminados. As CDMOs que fornecem serviços de localização única, combinadas com a experiência e um histórico de sucesso com formulações complexas, podem ajudar a superar muitos dos desafios atuais nesse campo. Como um desses CDMOs, a Ajinomoto Bio-Pharma Services continua a explorar processos inovadores de formulação de medicamentos, a fim de avançar no campo do desenvolvimento de medicamentos.