ICH Q12 Updates: Aumentar a previsibilidade e eficiência das alterações pós-aprovação
Fonte: GE Healthcare Life Sciences
Por Andrew Chang, Ph.D., Novo Nordisk Inc.
Este artigo foi criado a partir da apresentação do autor na conferência Bioprocessing Asia 2018, com a GE Healthcare como patrocinadora principal. A série BioProcessing Asia Conference foi criada para fornecer uma plataforma para avançar a contribuição das ciências de bioprocessamento para o desenvolvimento e fabricação de produtos biofarmacêuticos acessíveis na Ásia.
 A indústria farmacêutica de hoje está evoluindo rapidamente. Os avanços na ciência e tecnologia estão dando uma nova esperança para abordar as necessidades do paciente não satisfeitas com tratamentos altamente inovadores. No entanto, mesmo com esses desenvolvimentos inovadores no tratamento de pacientes, diversos ambientes regulatórios em todo o mundo criam encargos desnecessários no gerenciamento do ciclo de vida de produtos farmacêuticos. Isso inclui, mas não se limita a, alterações pós-aprovação feitas para aprimoramento contínuo ou expansão de recursos de fabricação para o fornecimento global de produtos. Esses encargos criam a necessidade de uma diretriz reguladora harmonizada, baseada em riscos, eficiente e previsível para o gerenciamento do ciclo de vida de produtos farmacêuticos.
A indústria farmacêutica de hoje está evoluindo rapidamente. Os avanços na ciência e tecnologia estão dando uma nova esperança para abordar as necessidades do paciente não satisfeitas com tratamentos altamente inovadores. No entanto, mesmo com esses desenvolvimentos inovadores no tratamento de pacientes, diversos ambientes regulatórios em todo o mundo criam encargos desnecessários no gerenciamento do ciclo de vida de produtos farmacêuticos. Isso inclui, mas não se limita a, alterações pós-aprovação feitas para aprimoramento contínuo ou expansão de recursos de fabricação para o fornecimento global de produtos. Esses encargos criam a necessidade de uma diretriz reguladora harmonizada, baseada em riscos, eficiente e previsível para o gerenciamento do ciclo de vida de produtos farmacêuticos.
Entre 2003 e 2012, a Conferência Internacional de Harmonização (ICH) desenvolveu suas diretrizes de Q8 a Q11 para enfatizar uma abordagem baseada na ciência e no risco para a qualidade farmacêutica. O ICH Q8 ao Q11 se concentrou principalmente nos estágios iniciais do ciclo de vida do produto, mas houve um processo significativo desde a sua criação. A ICH reconheceu a necessidade de uma nova diretriz focada na fase de fabricação comercial "para melhorar a execução e a comunicação da ciência e das avaliações baseadas em risco que permitem o gerenciamento do ciclo de vida do produto". 1O resultado foram as considerações técnicas e regulamentares da ICH Q12 para as diretrizes de gerenciamento do ciclo de vida de produtos farmacêuticos, que se baseiam nos princípios descritos nas diretrizes anteriores da ICH. O objetivo do Q12 é "demonstrar como o conhecimento aprimorado de produtos e processos contribui para uma redução no número de envios regulatórios pós-aprovação". 2
Seus objetivos incluíam, mas não se limitavam a:
- harmonizar o gerenciamento de mudanças de maneira mais transparente e eficiente nas regiões da ICH
- facilitando a supervisão regulatória baseada em risco
- enfatizando uma estratégia de controle como um componente-chave do dossiê regulatório
- promover a utilização de instrumentos de regulação para a gestão da mudança em potencial e permitindo a gestão estratégica das mudanças pós-aprovação.
Com a implementação adequada das ferramentas e capacitadores do Q12, o setor pode gerenciar efetivamente as alterações do CMC no sistema de qualidade farmacêutica de uma empresa, com menos necessidade de ampla supervisão regulatória antes da implementação.
Ferramentas reguladoras e ativadores ICH Q12
O grupo de trabalho de especialistas da ICH Q12 (EWG) era composto por quase 40 representantes regulatórios e do setor, com uma ampla gama de conhecimentos técnicos, de qualidade e regulatórios. Por exemplo, a equipe incluiu empresas que trabalham não apenas com produtos de pequenas moléculas, mas também com produtos biológicos. O grupo usou a estrutura existente estabelecida pela ICH Q8 a Q11, identificando ferramentas e facilitadores efetivos existentes, bem como novas que poderiam ajudar a alcançar os objetivos predefinidos do Q12 (Figura 1). Isso inclui aqueles usados apenas por países individuais, como categorias de relatórios baseados em risco para alterações pós-aprovação e protocolos de gerenciamento de alterações pós-aprovação (usados na Europa) e protocolos de comparabilidade (usados nos EUA).
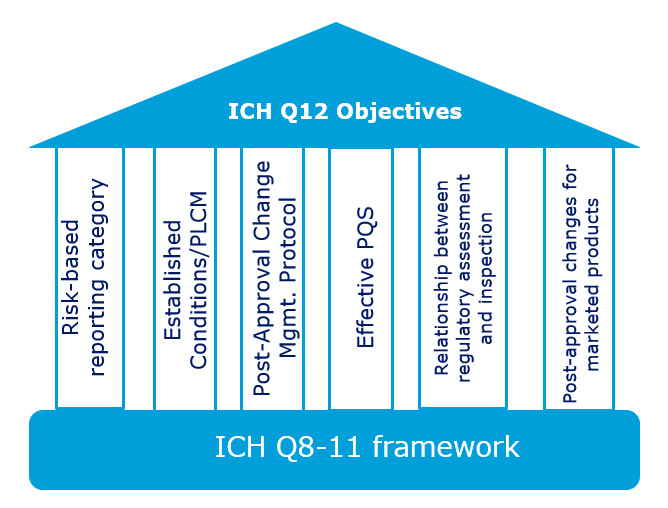
Figura 1: Ferramentas e capacitadores usados para atender aos objetivos predefinidos do ICH Q12
Mais informações sobre essas ferramentas podem ser encontradas no documento da Etapa 2b para ICH Q12. 3
ICH Q12 Condições estabelecidas
Condições estabelecidas (CEs) é uma nova ferramenta reguladora sob a estrutura da ICH que foi definida mais claramente nas diretrizes do Q12; Os CEs servem como informações juridicamente vinculativas consideradas necessárias para garantir a qualidade do produto.
Um aplicativo de marketing contém uma combinação de CEs e informações de suporte. As informações de suporte não são consideradas como CE, mas são fornecidas no aplicativo de marketing para compartilhar informações de desenvolvimento e fabricação em um nível adequado de detalhes com os reguladores e para justificar a seleção inicial de CEs e suas categorias de relatórios (Figura 2). As alterações feitas nas informações de suporte (por exemplo, mais conhecimento adquirido após a aprovação) não estão sujeitas a relatórios regulatórios.
Um aplicativo de marketing contém uma combinação de CEs e informações de suporte. As informações de suporte não são consideradas como CE, mas são fornecidas no aplicativo de marketing para compartilhar informações de desenvolvimento e fabricação em um nível adequado de detalhes com os reguladores e para justificar a seleção inicial de CEs e suas categorias de relatórios (Figura 2). As alterações feitas nas informações de suporte (por exemplo, mais conhecimento adquirido após a aprovação) não estão sujeitas a relatórios regulatórios.

Figura 2: Um exemplo de ECs versus informações de suporte.
O número de CEs e a definição restrita dos mesmos variarão com base em vários fatores, incluindo entendimento de produtos e processos, caracterização, abordagem de desenvolvimento de uma empresa e risco potencial à qualidade do produto. Por exemplo, se duas empresas trabalham na mesma classe de produto e uma delas possui menos conhecimento sobre um processo ou produto, elas podem acabar com mais CEs.
As seguintes abordagens durante o desenvolvimento do produto podem ser usadas sozinhas ou combinadas para identificar CEs:
- Uma abordagem baseada em parâmetros na qual o desenvolvimento do produto antes dos envios de regulamentação fornece um entendimento limitado do relacionamento entre insumos e atributos de qualidade resultantes. Ele incluirá um número maior de entradas (por exemplo, parâmetros do processo e atributos do material) junto com as saídas (incluindo controles em processo).
- Uma abordagem aprimorada com maior entendimento da interação entre entradas e atributos de qualidade do produto, juntamente com uma estratégia de controle correspondente, pode levar à identificação de CEs focados nos parâmetros de entrada mais importantes, juntamente com as saídas, conforme apropriado.
- Em certos casos, a aplicação do conhecimento de um ambiente rico em dados permite uma abordagem baseada no desempenho, na qual os ECs podem se concentrar principalmente no controle das saídas da operação da unidade, e não nas entradas do processo (por exemplo, parâmetros do processo e atributos do material).
A diretriz do Q12 também pretende aumentar a previsibilidade e definir expectativas apropriadas sobre quando os relatórios de alterações após a aprovação serão necessários com base nos CEs. O Q12 aconselha os titulares de Autorizações de Introdução no Mercado (AIM) a propor uma categoria de relatório em seu envio original ou em um suplemento pós-aprovação para produtos já comercializados, caso precisem fazer alterações futuras nos CEs. A categoria de relatório depende do risco potencial para a qualidade do produto. Isso cria um entendimento claro sobre quais mudanças nos CEs exigiriam supervisão regulatória e como elas devem ser relatadas. Os titulares de AIM podem seguir os regulamentos ou orientações regionais existentes ou propor uma categoria de relatório alternativa com justificativas. As atividades de avaliação de riscos devem seguir as abordagens descritas no ICH Q9,
A Figura 3 fornece um exemplo de uma árvore de decisão de uma categoria de relatório EC para um parâmetro de processo crítico / chave (CPP / KPP), conforme descrito no documento ICH Q12 Etapa 2b.
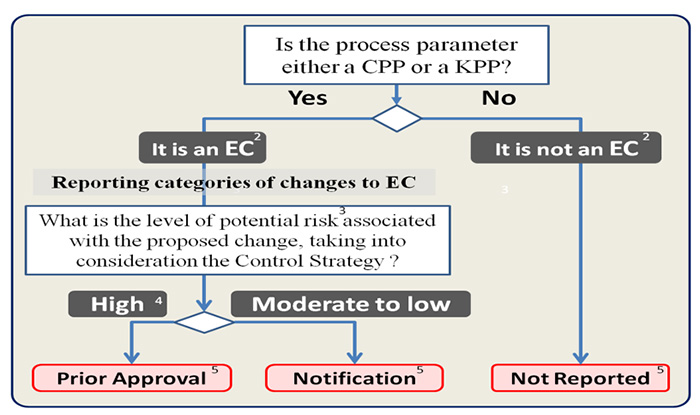
Figura 3: Árvore de decisão para a identificação de CEs e categorias de relatórios associadas aos parâmetros do processo de fabricação
A diretriz proposta na ICH Q12 alcançou a Etapa 2b no ano passado e foi publicada para comentários do público. O grupo de trabalho incentiva a indústria a expressar sua opinião enquanto a oportunidade está disponível. Com a implementação efetiva das ferramentas e capacitadores do Q12, os titulares da AIM aprimoram sua capacidade de gerenciar as alterações do CMC efetivamente em seu próprio sistema de qualidade, com menos necessidade de ampla supervisão regulatória antes da implementação. Esse marco reduziria encargos regulatórios desnecessários, diminuiria custos e incentivaria a inovação no crescente mercado atual.
- ICH, Documento conceitual final, Q12: considerações técnicas e regulamentares para o gerenciamento do ciclo de vida de produtos farmacêuticos - https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q12/Q12_Final_Concept_Paper_July_2014.pdf
- Considerações técnicas e regulamentares da FDA, Q12 para gerenciamento do ciclo de vida de produtos farmacêuticos - https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM609205.pdf
- Agência Europeia de Medicamentos, ICH Diretriz Q12 sobre considerações técnicas e regulamentares para gerenciamento do ciclo de vida de produtos farmacêuticos, Etapa 2b - https://www.ema.europa.eu/documents/scientific-guideline/draft-ich-guideline-q12-technical-regulatory -considerations-pharmaceutical-product-lifecycle_en.pdf
 O Dr. Andrew Chang tem mais de vinte anos de experiência no desenvolvimento, regulação e qualidade de produtos biológicos e farmacêuticos. Atualmente, como Vice-Presidente de Qualidade e Conformidade Regulatória, Inteligência e Inspeção da Qualidade, Novo Nordisk A / S, ele é responsável por assuntos externos, fornecendo consultoria e soluções estratégicas para questões relacionadas à qualidade e a regulamentação, além de suporte especializado para a preparação da inspeção. . Desde 2013, Andrew representa a Novo Nordisk em vários grupos de trabalho em organizações comerciais da indústria, por exemplo, PhRMA, BIO para defender os interesses dos pacientes e da indústria, desenvolvendo documentos de posição e participando de reuniões de contato com as autoridades reguladoras. Ele também é membro dos Grupos de Trabalho ICH da PhRMA e da BIO,
O Dr. Andrew Chang tem mais de vinte anos de experiência no desenvolvimento, regulação e qualidade de produtos biológicos e farmacêuticos. Atualmente, como Vice-Presidente de Qualidade e Conformidade Regulatória, Inteligência e Inspeção da Qualidade, Novo Nordisk A / S, ele é responsável por assuntos externos, fornecendo consultoria e soluções estratégicas para questões relacionadas à qualidade e a regulamentação, além de suporte especializado para a preparação da inspeção. . Desde 2013, Andrew representa a Novo Nordisk em vários grupos de trabalho em organizações comerciais da indústria, por exemplo, PhRMA, BIO para defender os interesses dos pacientes e da indústria, desenvolvendo documentos de posição e participando de reuniões de contato com as autoridades reguladoras. Ele também é membro dos Grupos de Trabalho ICH da PhRMA e da BIO,
Antes da Novo Nordisk, Andrew atuou por mais de onze anos no US FDA, mais recentemente como Diretor Associado de Política e Regulação, Vice-Diretor Interino e Cientista Regulatório Sênior da Divisão de Hematologia, Centro de Avaliação e Pesquisa em Biologia (CBER). Durante seu mandato, Andrew recebeu numerosos prêmios da FDA de alto nível por seu desempenho excepcional e excepcional em revisão e gerenciamento regulatórios, inspeção de BPF e desenvolvimento de políticas. Isso inclui, entre outros, a Menção Especial do Comissário da FDA por concluir com êxito a iniciativa da FDA sobre regulação da qualidade do produto e o Prêmio de Realização em Saúde Pública do CBER pelo excelente desempenho da revisão regulatória que resultou em evitar uma crise na disponibilidade do produto. Em 2002,
O treinamento científico formal de Andrew inclui pós-doutorado em imunologia pelo National Institutes of Health, Ph.D. em Bioquímica pela Universidade Estadual de Nova York e bacharel em Química Farmacêutica pela China Pharmaceutical University. Ele publicou numerosos artigos científicos revisados por pares em JAMA, J. Exp. Med., Blood, J. Imunol., Dev. Immunol. Thromb Haemost., Hemofilia, Engenharia Farmacêutica etc., e tem sido um orador frequente em conferências nacionais e internacionais.
Nenhum comentário:
Postar um comentário