Uma introdução aos planos de amostragem
Por Mark Durivage, Quality Systems Compliance LLC

Os planos de amostragem são amplamente utilizados em todas as organizações regulamentadas pelo FDA. A maioria das organizações possui um procedimento estatístico que especifica um certo nível de qualidade aceitável (AQL) com base no risco. (Caso contrário, eles deveriam!) No entanto, a maioria das pessoas segue os requisitos do procedimento sem compreender completamente como os planos de amostragem realmente funcionam.
A amostragem probabilística é baseada no fato de que cada membro de uma população tem a mesma chance de ser selecionado. Basicamente, os planos de amostragem estatística são usados para tomar decisões sobre a aceitação ou rejeição de produtos. Os planos de amostragem estatística são uma técnica de controle de qualidade comumente usada para inspeção de entrada, em andamento e final.
Os planos de amostragem são usados quando o teste é destrutivo e todas as peças seriam consumidas durante o teste, não deixando peças para uso ou distribuição comercial, o custo da inspeção de 100% é muito alto ou a inspeção de 100% leva muito tempo.
Os planos de amostragem são um meio de identificar, não impedir, a baixa qualidade. Os planos de amostragem não substituem o controle do processo .
Os planos de amostragem podem ser usados para dados variáveis e de atributos. Lembre-se de que os dados variáveis podem ser medidos em uma escala contínua e os dados de atributos medem pontos de dados discretos, como aprovação / reprovação, aprovação / não aprovação, e coisas que podem ser contadas.
Todas as amostras estão sujeitas a riscos
Toda a amostragem estatística está sujeita a risco. Os riscos são definidos pela margem de erro e intervalos de confiança. Idealmente, um plano de amostragem deve rejeitar todos os lotes "ruins" e aceitar todos os lotes "bons". No entanto, como os planos de amostragem baseiam as decisões em uma amostra do lote e não no lote inteiro, sempre há uma chance de tomar uma decisão incorreta. Esses erros são chamados de risco do produtor e risco do consumidor. Erro Alfa Tipo I (α) O risco do produtor é a probabilidade de rejeitar um bom lote. Erro beta do tipo II (β) O risco do consumidor é a probabilidade de aceitar um lote ruim.
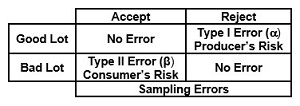
Figura 1: Risco do produtor e do consumidor
Como funcionam os planos de amostragem
Todos os planos de amostragem utilizam o conceito de lote (N), amostra (n) retirada aleatoriamente do lote e um número de aceitação (c). Muito é um todo - todos os membros de um grupo - e uma amostra é um subconjunto de muito. Para cada lote inspecionado, tomamos uma decisão de aceitação / rejeição com os lotes aceitos liberados para o estoque e os lotes rejeitados segregados, rotulados e colocados em quarentena.
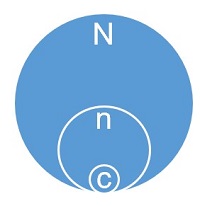
Figura 2: Representação gráfica de um lote (N), amostra (n) e número de aceitação (c)
Por exemplo, um lote contendo 2.500 (N) peças é inspecionado usando um AQL de 1.0, que exige que 42 (n) peças sejam selecionadas aleatoriamente no lote e inspecionadas. Neste exemplo, o número de aceitação é 0 (c). Se todas as 42 peças selecionadas aleatoriamente são inspecionadas e todas as peças atendem aos critérios de inspeção (zero falhas), o lote é aceito (passa). No entanto, se houver uma falha, o lote é rejeitado (falha) e deve ser segregado, rotulado e colocado em quarentena até ser tratado pelo MRB (Material Review Board).
Curva característica de operação
O comportamento de um plano de amostragem é descrito graficamente pela curva característica operacional do plano de amostragem (OCC). As curvas de características operacionais são geradas usando a distribuição binomial ou Poisson, com exceção dos planos de amostragem C = 0. A distribuição hipergeométrica é usada para gerar a curva da característica operacional para planos de amostragem C = 0. As curvas podem ser facilmente construídas usando tabelas de referência, calculadoras ou planilhas.
A curva de característica operacional mostra a probabilidade de aceitar lotes de diferentes níveis de qualidade para um plano de amostragem específico e ajuda a discriminar entre lotes bons e ruins. A forma exata e a localização da curva são definidas pelo tamanho da amostra (n) e número de aceitação (c) para o plano de amostragem.
O nível de qualidade aceitável (AQL) é uma medida do nível de qualidade rotineiramente aceito por esse plano de amostragem. É definido como o percentual de defeito que o plano de amostragem aceitará 95% do tempo. Isso significa que lotes iguais ou superiores ao AQL são aceitos pelo menos 95% das vezes e rejeitados no máximo 5% das vezes. O AQL pode ser determinado usando o OCC, encontrando o nível de qualidade no eixo inferior que corresponde a uma probabilidade de aceitação de 0,95 (95%) no eixo esquerdo (veja a Figura 3).
O percentual de tolerância ao lote com defeito (LTPD) é o nível de qualidade rotineiramente rejeitado pelo plano de amostragem. Geralmente, é definido como o percentual de defeito que o plano de amostragem rejeitará 90% do tempo. Em outras palavras, esse também é o percentual de defeito que será aceito pelo plano de amostragem no máximo 10% do tempo. Isso significa que lotes iguais ou inferiores ao LTPD são rejeitados pelo menos 90% das vezes e aceitos no máximo 10% das vezes. O LTPD pode ser determinado usando o OCC, encontrando o nível de qualidade no eixo inferior que corresponde a uma probabilidade de aceitação de 0,10 (10%) no eixo esquerdo (veja a Figura 3).
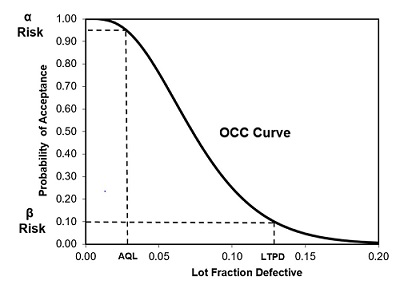
Figura 3: Exemplo de curva de característica operacional (OCC)
O número de aceitação (c) tem um impacto significativo na forma do OCC. Como mostra a Figura 4, à medida que o número de aceitação (c) aumenta, o risco do produtor (α) diminui e, inversamente, à medida que o número de aceitação (c) diminui, o risco do produtor (α) aumenta. Além disso, à medida que o número de aceitação (c) aumenta, o risco do consumidor (β) aumenta e, inversamente, à medida que o número de aceitação (c) diminui, o risco do consumidor (β) diminui. Os planos de amostragem C = 0 desenvolvidos por Nicholas Squeglia são amplamente aceitos nas indústrias regulamentadas pela FDA, pois proporcionam maior proteção ao consumidor.

Figura 4: Exemplo de curva característica característica (OCC) para C = 0, C = 3 e C = 5
Um OCC ideal / perfeito não produzirá nenhum Erro Tipo I (α) Risco do Produtor ou Erro Tipo II (β) Risco do Consumidor para uma determinada fração de lote com defeito ou menos.
O OCC mostrado na Figura 5 é considerado ideal / perfeito. Para esta curva, uma fração com defeito de 0,25 libras será aceita 100% do tempo. Na realidade, o CCO ideal / perfeito não existe.
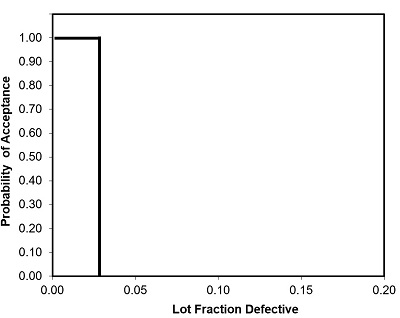
Figura 5: Curva teórica de característica operacional perfeita (OCC)
Conclusão
Os planos de amostragem devem estar ligados ao risco. Deveria ser óbvio que mais inspeção é necessária e deve ser realizada para itens de maior risco e menos amostragem deve ser necessária e realizada para itens de menor risco. Certifique-se de que seu programa de treinamento forneça algumas informações sobre como e por que os planos de amostragem funcionam para aqueles que realizam atividades de amostragem.
Vale a pena repetir: os planos de amostragem são um meio de identificar, e não impedir, a baixa qualidade. Os planos de amostragem não substituem o controle do processo . Um programa de amostragem eficaz pode ser usado para aumentar e apoiar o controle do processo.
Referências:
- ANSI / ASQ Z1.4-2008: Procedimentos de amostragem e tabelas para inspeção por atributos
- Procedimentos e tabelas de amostragem ANSI / ASQ Z1.9-2003 (R2013) para inspeção por variáveis para porcentagem não conforme
- Durivage, MA, 2014, Estatísticas práticas de engenharia, processos e confiabilidade , Milwaukee, ASQ Quality Press
- Squeglia, Nicholas K. 2008, Planos de Amostragem com Número de Aceitação Zero. 5a ed. Milwaukee: ASQ Quality Press.
Sobre o autor:
 Mark Allen Durivage trabalhou como médico, educador, consultor e autor. Ele está gerenciando o consultor principal da Quality Systems Compliance LLC, um membro da ASQ e um membro da SRE. Ele obteve um BAS em usinagem auxiliada por computador pela Universidade de Siena Heights e um MS em gerenciamento de qualidade pela Eastern Michigan University. Ele possui várias certificações, incluindo CRE, CQE, CQA, CSQP, CSSBB, RAC (Global) e CTBS. Ele escreveu vários livros disponíveis na ASQ Quality Press, publicou artigos no Quality Progress e é colaborador frequente do Life Science Connect. Durivage reside em Lambertville, Michigan. Não hesite em enviar um e-mail para mark.durivage@qscompliance.com ou conectar-se a ele no LinkedIn .
Mark Allen Durivage trabalhou como médico, educador, consultor e autor. Ele está gerenciando o consultor principal da Quality Systems Compliance LLC, um membro da ASQ e um membro da SRE. Ele obteve um BAS em usinagem auxiliada por computador pela Universidade de Siena Heights e um MS em gerenciamento de qualidade pela Eastern Michigan University. Ele possui várias certificações, incluindo CRE, CQE, CQA, CSQP, CSSBB, RAC (Global) e CTBS. Ele escreveu vários livros disponíveis na ASQ Quality Press, publicou artigos no Quality Progress e é colaborador frequente do Life Science Connect. Durivage reside em Lambertville, Michigan. Não hesite em enviar um e-mail para mark.durivage@qscompliance.com ou conectar-se a ele no LinkedIn .